
Abstract
Aims: The MeRes-1 trial sought to study the safety and effectiveness of a novel sirolimus-eluting bioresorbable vascular scaffold (MeRes100 BRS) in treating de novo native coronary artery lesions by clinical evaluation and using multiple imaging modalities.
Methods and results: The MeRes-1 first-in-human trial was a single-arm, prospective, multicentre study, which enrolled 108 patients with de novo coronary artery lesions (116 scaffolds were deployed to treat 116 lesions in 108 patients). At six months, quantitative coronary angiography revealed in-scaffold late lumen loss of 0.15±0.23 mm with 0% binary restenosis. Optical coherence tomography demonstrated minimum scaffold area (6.86±1.73 mm2) and percentage neointimal strut coverage (99.30%). Quantitative intravascular ultrasound analysis confirmed a 0.14±0.16 mm2 neointimal hyperplasia area. At one year, major adverse cardiac events, a composite of cardiac death, any myocardial infarction and ischaemia-driven target lesion revascularisation, occurred in only one patient (0.93%) and there was no scaffold thrombosis reported. At one year, computed tomography angiography demonstrated that all scaffolds were patent and in-scaffold mean percentage area stenosis was 11.33±26.57%.
Conclusions: The MeRes-1 trial demonstrated the safety and effectiveness of MeRes100 BRS. The favourable clinical outcomes and effective vascular responses have provided the basis for further studies in a larger patient population. The MeRes-1 trial is registered at the Clinical Trials Registry-India. CTRI number: CTRI/2015/04/005706
Abbreviations
BRS: bioresorbable vascular scaffolds
CTA: computed tomography angiography
IVUS: intravascular ultrasound
OCT: optical coherence tomography
QCA: quantitative coronary angiography
Introduction
Bioresorbable vascular scaffolds (BRS) may offer significant advantages over metallic stents in the treatment of coronary artery disease1. Several biomaterials have been developed and are being investigated for clinical application as BRS: these include polymers such as poly-L-lactic acid (PLLA), poly(desaminotyrosil-tyrosine ethyl ester) carbonate, polycaprolactone, salicylic acid and metals such as magnesium and iron2. Amongst these, PLLA remains the most commonly used polymer because of good biocompatibility, biodegradability, a high mechanical strength and good shaping and moulding properties3.
Among the present generation of commercially available BRS, the Absorb™ bioresorbable vascular scaffold (Abbott Vascular, Santa Clara, CA, USA) has structural and mechanical limitations that restrict its extensive application in “real-world” interventional practice. The greater strut thickness (156 µm) and profile limit its deliverability. The large struts also create laminar flow disruptions and delayed endothelialisation, which could potentially be responsible for increased scaffold thrombosis. Modifications are needed in the scaffold technology to overcome these limitations4. It is desirable for the next-generation BRS to possess reduced strut thickness and profile to enhance deliverability. This may potentially result in predictive endothelialisation, resorption and lower scaffold thrombosis.
The MeRes100™ sirolimus-eluting bioresorbable vascular scaffold system (Meril Life Sciences Pvt. Ltd., Vapi, India) is based on a biocompatible and bioresorbable polymer PLLA with a strut thickness of 100 µm. In the first-in-human MeRes-1 clinical trial, we have studied the new scaffold for safety and effectiveness using multimodality imaging and clinical outcomes at six-month and one-year follow-up.
Methods
STUDY DESIGN
The MeRes-1 is a prospective, multicentre, single-arm, open-label clinical trial of the MeRes100 sirolimus-eluting bioresorbable vascular scaffold system (MeRes100 BRS) for treating de novo native coronary artery lesions. The study was conducted at 13 Indian sites. It was performed in compliance with ICH-GCP guidelines, the Declaration of Helsinki and with the approval of the local institutional ethics committees. All patients provided written informed consent for trial participation.
STUDY POPULATION
A total of 108 patients were enrolled in the study between May 2015 and April 2016. We included patients aged 18-65 years with up to two de novo lesions in native coronary arteries (one lesion per target vessel) with reference vessel diameters of 2.75-3.50 mm and lesion length ≤20 mm. The patients presented with stenosis >50% and <100% with a Thrombolysis In Myocardial Infarction (TIMI) flow grade of >1.
Key exclusion criteria were patients with acute myocardial infarction (MI) <7 days, those with prior revascularisation by either bypass surgery or percutaneous coronary intervention (PCI), left ventricular ejection fraction ≤30%, left main lesions, aorto-ostial lesions, moderate to severe calcification at the lesion site, bifurcation with a side branch >2 mm in diameter with ostial disease, arterial/vein graft lesions and severe angulation and tortuosity of the target vessel and total occlusions.
STUDY DEVICE DESCRIPTION
The MeRes100 BRS is a balloon-expandable PLLA polymer backbone scaffold. The top contains an active drug coating of sirolimus distributed at a dose of 1.25 μg/mm2 formulated in a 1:1 mixture of biocompatible and bioabsorbable polymer poly-D, L-lactide (PDLLA), which acts as a drug reservoir and controls the drug release rate. The thin uniform coating is 3-4 µm and does not web, crack or lump, as studied by scanning electron microscopy (data on file). Both PLLA and PDLLA undergo hydrolytic degradation of the ester bonds in the polymers, generating lactic acid which is converted to CO2 and H2O which gets eliminated from the body. The expected degradation of the scaffold from the treatment site is within 24-36 months of implantation (data on file).
The MeRes100 BRS has a hybrid cell design, close cells at the edges and open cells along the length (Figure 1), which ensures optimal vessel wall conformability. It has a low strut thickness of 100 µm and the strut width varies from 150-200 µm, depending on scaffold diameters. The crossing profile is 1.20 mm and 1.25 mm for 3.0 mm and 3.5 mm diameters, respectively. The couplets of tri-axial platinum radiopaque markers fixed circumferentially 120º apart from each other at either end of the scaffold allow convenience of viewing the scaffold in two orthogonal views during its deployment. For the study, MeRes100 BRS in lengths of 19 and 24 mm and diameters of 2.75, 3.00 and 3.50 mm were used.
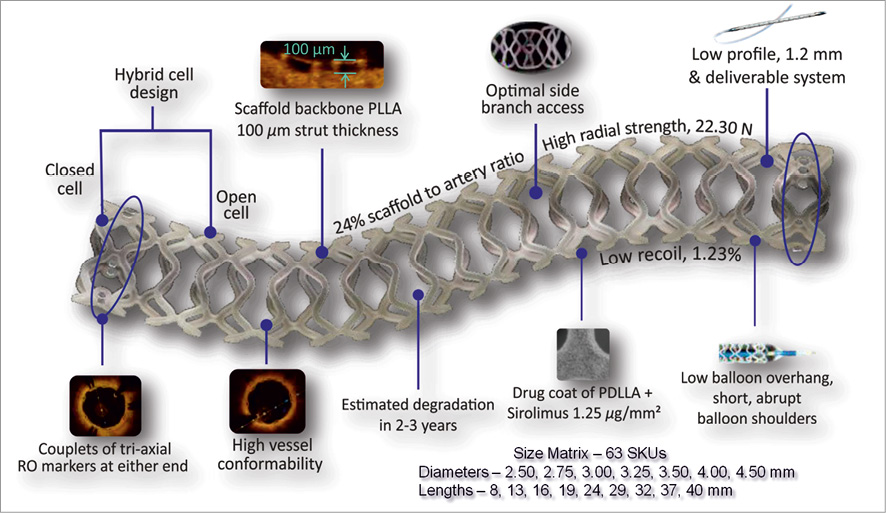
Figure 1. Design of the MeRes100 bioresorbable vascular scaffold stent platform.
In preclinical studies utilising OCT and histopathology, the MeRes100 showed equivalent in vivo acute and chronic recoil, as well as similar neointimal formation and arterial healing when compared to the benchmark Absorb BVS up to 180 days. Preliminary evaluation of serial OCT obtained at one and two years suggests a more gradual integration of the scaffold into the arterial wall than that reported for Absorb (2% of preserved box appearance in MeRes100 vs. 80.4% for Absorb at two years5).
INTERVENTIONAL PROCEDURE
The target lesions were treated according to standard guidelines for the PCI procedure6. For MeRes100 BRS implantation, predilatation was mandatory, and it was recommended to use a predilatation balloon which was either 0.5 mm smaller in diameter or at least 1:1 sized with the reference vessel diameter. The expansion of the predilatation balloon was maintained up to its nominal pressure. After scaffold deployment, high-pressure post-dilatation at ≥18 atmospheres was mandatory with either an optimal balloon size or a 0.5 mm larger non-compliant balloon to achieve residual diameter stenosis of ≤10%. Full lesion coverage with at least 2 mm on either side of the target lesions was recommended with the MeRes100 BRS. Dual antiplatelet therapy with aspirin 75-150 mg/day and clopidogrel 75 mg/day or prasugrel 10 mg/day or ticagrelor 90 mg/day was maintained for a minimum duration of one year, beyond which a switch to single antiplatelet therapy with aspirin alone was left to the operator’s discretion.
STUDY ENDPOINTS
The major safety primary endpoints were major adverse cardiac events (MACE) - a composite of cardiac death, any MI, and ischaemia-driven target lesion revascularisation (ID-TLR) - at six months and one year. Ischaemia-driven TLR was defined by a diameter stenosis (DS) of ≥50% with ischaemia or symptoms, or a diameter stenosis of ≥70% observed even without any signs and symptoms of ischaemia at the time of follow-up angiography. The World Health Organization definition for periprocedural MI was used, which was defined as PCI-related, i.e., three times (3x) higher cardiac biomarkers, and CABG-related, i.e., five times (5x) greater cardiac biomarker values than the 99th percentile upper reference limit. The major safety secondary endpoints were device and procedural success and scaffold thrombosis (Academic Research Consortium definition)7 at six months and one year. Device success was defined as successful deployment of the scaffold in the intended target lesion with a residual stenosis <20% by quantitative coronary angiography (QCA). Procedural success was defined as achieving angiographic success without MACE during hospitalisation.
At six-month follow-up, the major effectiveness endpoints were in-segment and in-scaffold late lumen loss (LLL) using QCA, minimal lumen area and neointimal hyperplasia (NIH) area with optical coherence tomography (OCT), and scaffold and lumen area by intravascular ultrasound (IVUS). Also, at 12-month follow-up mean lumen area was determined using computed tomography angiography (CTA) imaging.
All clinical endpoint events were adjudicated by an independent clinical events committee to minimise the impact of possible bias on clinical outcomes. The cumulative safety data from the trial at pre-specified intervals were reviewed by the data and safety monitoring board.
MULTIMODALITY IMAGING ASSESSMENTS
The QCA was analysed by the Cardiovascular Research Centre, Sao Paulo, Brazil. The treated segment and peri-scaffold segment were evaluated using offline QCA at the post-index procedure and six-month follow-up. The computed QCA parameters were analysed with dedicated QAngio XA software, version 7.3 (Medis, Leiden, the Netherlands).
The IVUS, OCT, and CTA analyses were performed at Cardialysis BV, Rotterdam, the Netherlands. The OCT images were acquired using a C7 Dragonfly™ and C7-XR™, Fourier-Domain OCT system (LightLab Imaging [now St. Jude Medical], Westford, MA, USA), incorporating a pullback inside the deployed scaffold. Computed tomography angiography imaging was performed using 128-slice dual-source CT, Definition Flash (Siemens, Munich, Germany). The CTA outcomes of 12 patients were analysed at one year post index procedure using quantitative CTA (TeraRecon, Foster City, CA, USA). For the evaluation, the presence and extent (number of segments) of specific morphologic characteristics of plaques were recorded as calcified plaque, mixed plaque, and non-calcified plaque. The mean % area stenosis was calculated as: (reference lumen area minus in-scaffold minimal lumen area divided by reference lumen area) multiplied by 100. Reference lumen area was calculated as the mean of the 5 mm distal and proximal lumen area ignoring cross-sections of the two edges.
STATISTICAL ANALYSIS
All clinical, angiographic, IVUS, OCT and CTA endpoints were analysed based on per-treatment evaluable patient (PTE) populations. Categorical variables are expressed as frequency and percentages. Continuous variables are expressed as mean, standard deviation and 95% confidence intervals calculated using a Gaussian approximation. Paired comparisons of continuous variables between baseline and follow-up were calculated with the Student’s t-test. The Shapiro-Wilk statistic was calculated and a normal distribution was assumed when the p-value exceeded 0.05. SAS, version 9.3 (SAS Institute, Cary, NC, USA) was used for all statistical analysis.
Results
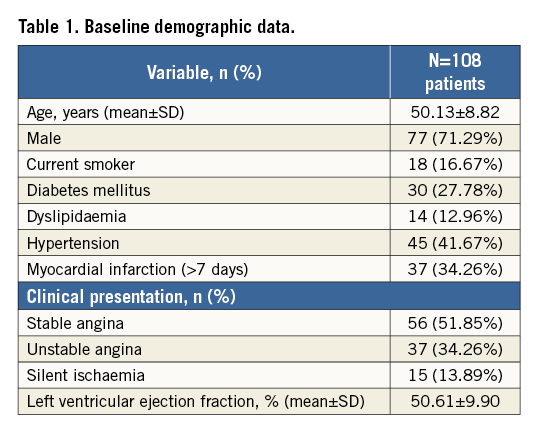
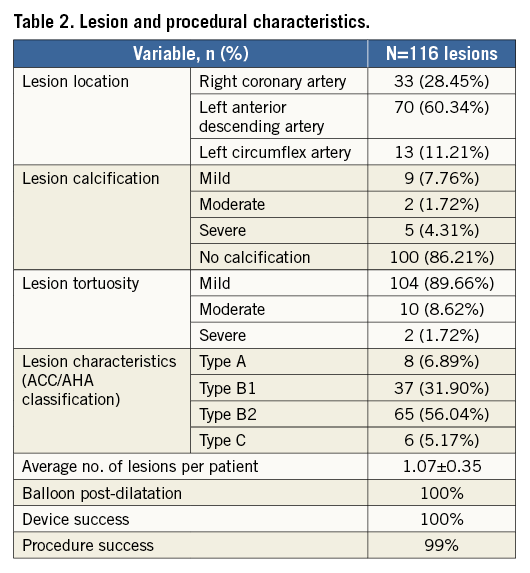
The MeRes-1 trial enrolled 108 patients with a mean age of 50.13±8.82 years. The baseline demographic data of the patients are given in Table 1. Procedural success was 99%, as one patient required bail-out stenting with metallic DES because of a proximal dissection during post-dilatation. Lesion and procedural characteristics are summarised in Table 2.
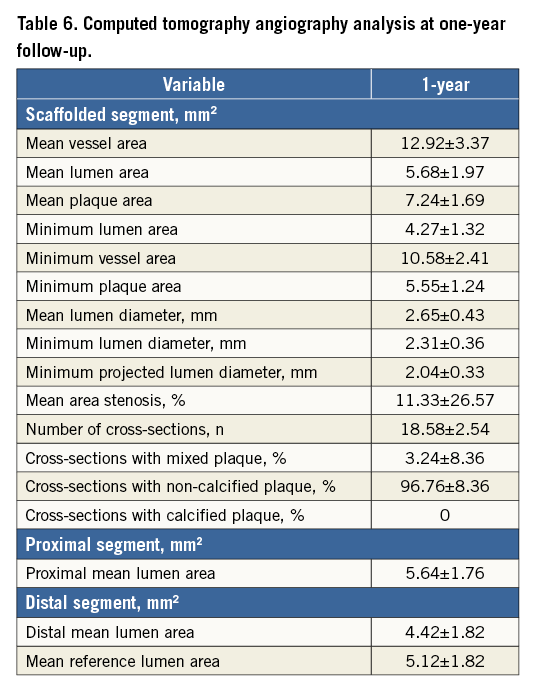
The baseline, post-index procedure, and six-month follow-up outcomes of QCA analysis are summarised in Table 3. At follow-up, in-scaffold mean lumen diameter reduced from 2.82±0.32 to 2.67±0.40 mm. The mean in-scaffold angiographic LLL was 0.15±0.23 mm (Figure 2). The cumulative frequency distribution curve of in-scaffold LLL up to six months is illustrated in Figure 3. The angiographic appearance pre-procedure, post-procedure and at six-month follow-up in a patient treated with MeRes100 BRS is presented in Figure 4. Serial quantitative IVUS analysis of 12 patients showed a low percentage volume obstruction of 2.53±2.95% with an NIH area of 0.14±0.16 mm2 at six months (Table 4). The mean scaffold area was maintained at six-month follow-up with no recoil. The OCT findings are summarised in Table 5. Neointimal coverage of struts was almost complete (99.30±1.38%) with a homogeneous pattern at six months. An example of a characteristic six-month OCT appearance is included in Figure 5. At one-year follow-up, 12 patients underwent CTA analysis and the results obtained are summarised in Table 6. The mean percentage area stenosis was 11.33±26.57%, which demonstrated favourable lumen patency.
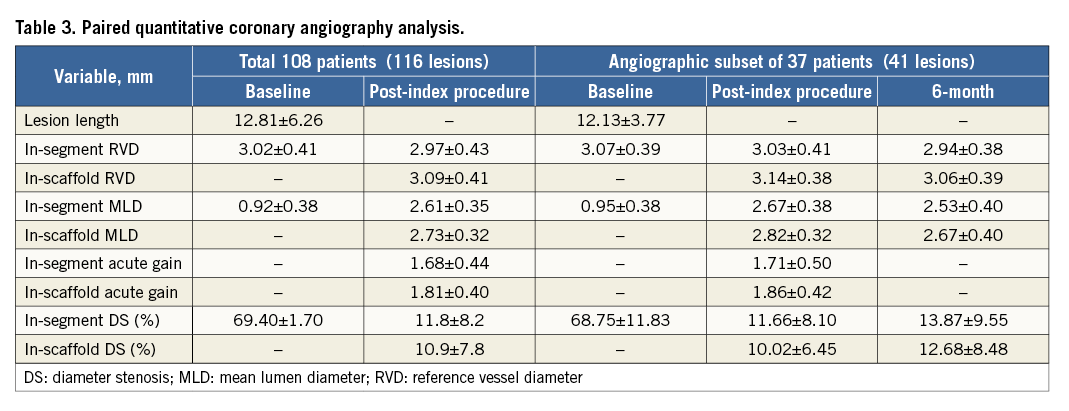
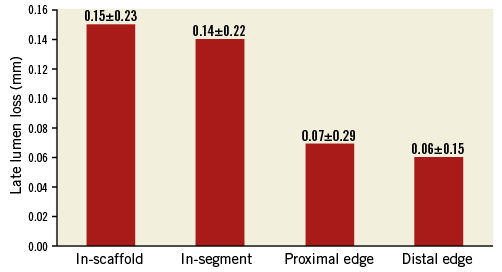
Figure 2. Late lumen loss at six-month follow-up.
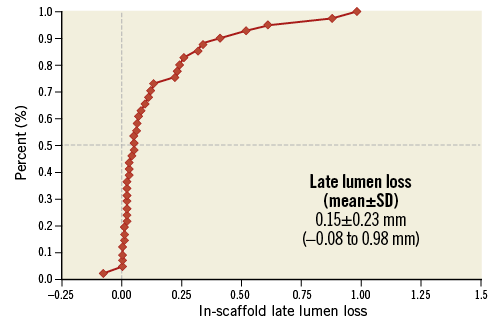
Figure 3. Cumulative frequency distribution curve for late lumen loss at six-month follow-up.
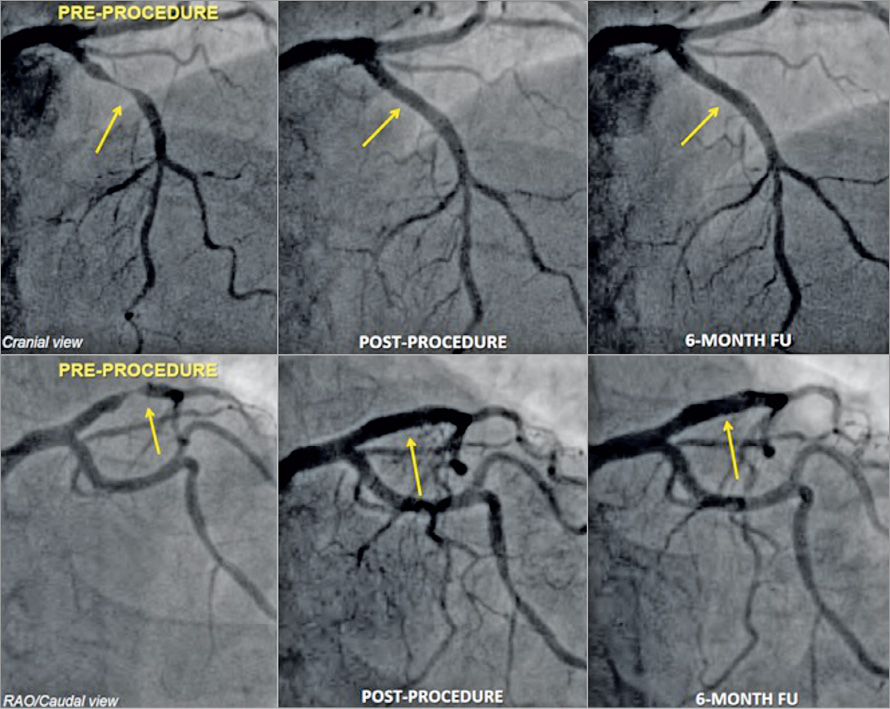
Figure 4. Angiographic depiction of pre-, post-procedure and six-month follow-up.
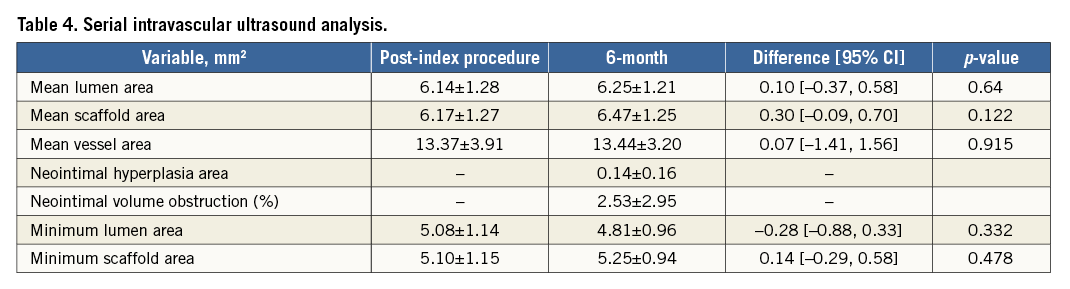
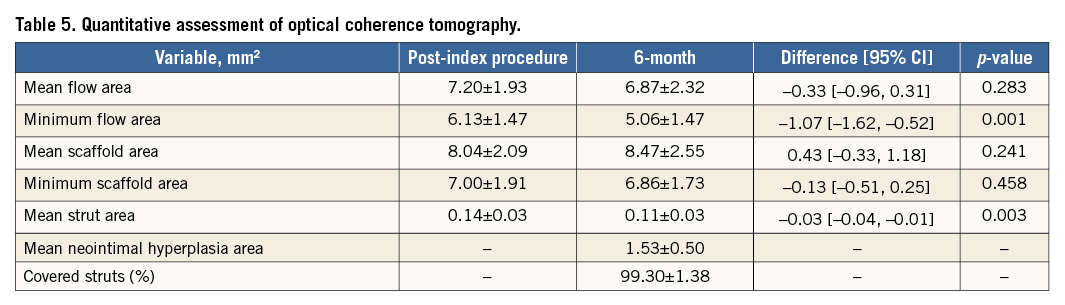
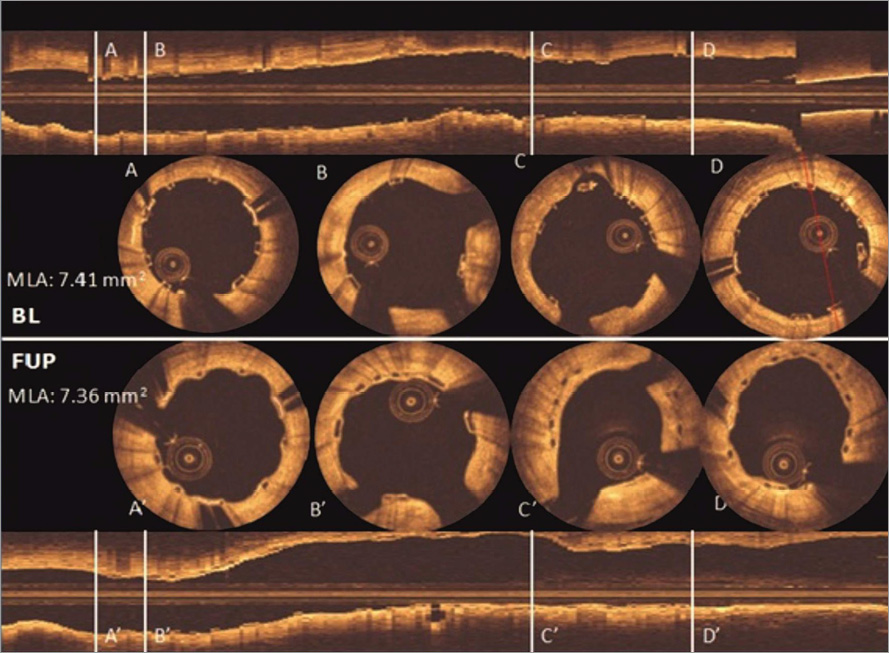
Figure 5. Post-implantation and six-month follow-up OCT images. The struts are observed as preserved boxes in the baseline images while the follow-up images show open boxes because of scaffold dissolution. At six-month follow-up, the neointimal coverage of the struts is presented as the membranous bridging pattern between the open boxes.
At six-month clinical follow-up, there were no MACE. One non-cardiac death occurred at five months due to aminophylline-induced anaphylactic shock. One case of ID-TLR (0.93%) was observed between six months and one year. No scaffold thrombosis (0%) was observed during the study up to one year.
Discussion
The MeRes-1, first-in-human trial of MeRes100 next-generation, low strut thickness BRS demonstrates a high device success rate, low MACE rate, no scaffold thrombosis and favourable imaging evaluation up to one year. Subgroup angiographic analysis showed an absence of in-scaffold and in-segment binary restenosis and late scaffold recoil. The IVUS subset analysis showed a non-significantly enlarged scaffold area between the index procedure and six months, and OCT demonstrated near complete neointimal strut coverage (99.3%). The CTA at one year showed maintained luminal patency. As a result, the MeRes100 BRS possessed favourable safety and effectiveness parameters in patients with de novo non-complex coronary artery lesions at one-year follow-up.
Currently available BRS have mechanical and structural constraints that may prevent their widespread use in real-world intervention and may also be predisposed to increased complications. Absorb BVS technology was introduced with relatively bulky struts including polymer-drug coating (156 µm) which are prone to restrict deliverability and create laminar flow disruption, leading to a higher TLR rate of 4.0%, 1.2%, 3.0%, 2.6% and 2.5% in the ABSORB Cohort B, ABSORB II, ABSORB III, ABSORB Japan and ABSORB China clinical trials, respectively, at one-year follow-up8-11. The strut thickness of 120 µm of the DREAMS scaffold (Biotronik, Bülach, Switzerland) and 150 µm of the DESolve® bioresorbable coronary scaffold (BCS) (Elixir Medical Corporation, Sunnyvale, CA, USA) demonstrated a higher TLR rate of 6.5% and 6.7%, respectively, at one year1,12. The MeRes100 BRS is engineered with thinner struts (100 µm), which predictably results in a mere 0.93% of ID-TLR at one-year follow-up.
Moreover, scaffold recoil was observed with the first version of the Absorb BVS device. The ABSORB Cohort A trial evaluated 30 patients treated with the scaffold for de novo coronary artery lesions. Scaffold recoil was evident from the high LLL of 0.44 mm and percentage in-scaffold diameter stenosis of 27% observed with QCA analysis at six months. Both these results were higher than those observed in the current study (LLL: 0.15±0.23 mm, and %DS: 12.68±8.48). The ABSORB study identified that the BVS was not able to withstand the vascular negative remodelling forces and thus led to scaffold recoil13. These limitations prompted modifications in the scaffold design. The second-generation Absorb BVS was evaluated in the ABSORB Cohort B trial. While the redesigned scaffold with increased radial and mechanical strength was able to limit LLL to 0.19±0.18 mm at six months, a case of strut fracture was noted on post-dilatation, indicating shortcomings in the scaffold design. Furthermore, the IVUS examination identified a non-significant reduction in the mean lumen area (6.53±1.24 to 6.36±1.18 mm2) and mean scaffold area (6.53±1.23 to 6.42±1.17 mm2) at six months. However, with MeRes100 lumen enlargement at six months was evidenced by a non-significant increase in mean lumen area and mean scaffold area by IVUS (mean lumen area: 6.14±1.28 to 6.25±1.21 mm2 and mean scaffold area: 6.17±1.27 to 6.47±1.25 mm2) and OCT (mean scaffold area: 8.04±2.09 to 8.47±2.55 mm2). The difference in minimal lumen area in ABSORB Cohort B (33 patients) was –0.32±0.62 at six months, which is similar to the MeRes100 (δ; –0.28 mm) in 12 patients14.
An increase in the mean scaffold area was observed with the DESolve BCS in the first-in-human trial involving 15 patients12. These findings were analogous in magnitude to the MeRes100 BRS. Comparison of outcomes at six months including LLL (DESolve BCS: 0.19±0.19 mm vs. MeRes100 BRS: 0.15±0.23 mm), IVUS-derived percentage neointimal volume obstruction (DESolve BCS: 7.18±3.37 vs. MeRes100 BRS: 2.53±2.95), QCA-derived in-scaffold percentage diameter stenosis (DESolve BCS: 12.63±11.37 vs. MeRes100 BRS: 12.68±8.48), and OCT-identified percentage strut coverage (BCS: 98.68±2.44 vs. MeRes100 BRS: 99.30±1.38) suggests a favourable safety and effectiveness profile for the MeRes100 BRS. Moreover, CTA outcomes confirmed vessel patency at one-year follow-up. In-scaffold MLD (2.67±0.40 mm) at six months by QCA analysis was identical with the 2.65±0.43 mm of CTA at one-year follow-up, which is a reflection of the good mechanical support provided by the MeRes100 BRS over the period of one year. Also, a non-significant difference was found between the minimum lumen area at six-month follow-up (4.81±0.96 mm2) by QCA analysis and one-year follow-up (4.27±1.32 mm2) by CTA analysis, which suggests that there was no scaffold recoil over the period of one year. The CTA results of mean lumen area for the Absorb BVS at 18-month and DESolve at 12-month follow-up were 5.2±1.3 mm2 (median of 4.94 mm2; 3.83 to 6.30) and 6.7±2.5 mm2, respectively which is comparable to MeRes100 BRS (5.68±1.97 mm2) at one-year follow-up. Moreover, in-scaffold percentage area stenosis of the Absorb BVS at 18-month follow-up was 34.0±15.0% (median of 23.2%; 11.42 to 32.55) and median of 16.9% (3.33 to 29.91) at 72 months15,16, while the MeRes100 BRS showed only 11.33±26.57% by CTA analysis at one year, which indicates that vessel patency was maintained following the index procedure.
The failure of the bioresorbable scaffold is usually due to the device’s inability to withstand the biomechanical challenges imposed by neointimal growth, atherosclerotic plaque, scaffold recoil and scaffold thrombosis. Variable factors including the selection of antirestenotic drug, strut thickness and type of biodegradable polymer play a vital role in the safety and effectiveness of any coronary device18. The MeRes100, which is a new generation of BRS with an innovative architecture, improved radiopacity, lower profile and thinner struts, demonstrates low LLL (0.15±0.23 mm), low ID-TLR (0.93%) and maintained vessel patency at one-year follow-up.
Study limitations
The study has the following limitations.
a. It was a single-arm study with a small sample size.
b. Relatively simple lesions were included in the study.
c. Although the clinical follow-up was 100%, only a small number of predefined patient populations underwent QCA, IVUS, OCT and CTA evaluation. These combined observations of quantitative evaluation by different imaging modalities have been encouraging. On the other hand, this is the first-in-human study of a novel “first of its type” thin-strut scaffold to establish its proof of concept, safety and effectiveness through multiple imaging modalities with the intention of moving forward with the device into a larger clinical evaluation.
Conclusions
The study supports the fact that MeRes100 BRS is safe and effective in simple, de novo coronary artery lesions. The vascular responses observed by OCT, IVUS and CTA are encouraging and comparable to metallic DES. These favourable results provide the basis for further randomised study in a larger patient population with longer follow-up.
| Impact on daily practice The present study demonstrated the safety and effectiveness of the novel PLLA-based sirolimus-eluting thin-strut MeRes100 BRS. The favourable clinical outcomes and the vascular responses observed at six-month and one-year follow-up provide the basis for a larger, randomised trial against a second-generation metallic drug-eluting coronary stent. The hope is that the second-generation BRS will overcome some of the technical limitations of first-generation BRS and bring benefit to more patients who have coronary artery disease. |
Guest Editor
This paper was guest edited by Davide Capodanno, MD, PhD; Cardio-Thoracic-Vascular Department, Ferrarotto Hospital, University of Catania, Catania, Italy.
Funding
Meril Life Sciences Pvt. Ltd. is the sponsor of the MeRes-1 trial.
Conflict of interest statement
A. Seth and A. Abizaid are external Scientific Advisors to Meril Life Sciences Pvt. Ltd. for clinical trial studies. A. Seth is also the Principal Investigator for the MeRes-1 study. The other authors have no conflicts of interest to declare. The Guest Editor has no conflicts of interest to declare.
