
Abstract
Aims: The aim of this study was to investigate clinical outcomes of patients at high risk of restenosis after implantation of a bioresorbable vascular scaffold (BVS).
Methods and results: The COMPARE-ABSORB trial was an investigator-initiated, prospective randomised study. Patients at high risk of restenosis were randomly assigned to receive either a BVS or an everolimus-eluting stent (EES). A dedicated implantation technique was recommended for BVS. The primary endpoint was target lesion failure (TLF), defined as the composite of cardiac death, target vessel myocardial infarction (TVMI) or clinically indicated target lesion revascularisation at one year. The enrolment was discontinued prematurely because of a high thrombosis and TVMI rate in the BVS arm. A total of 1,670 patients were recruited (BVS 848 patients and EES 822 patients). TLF occurred in 43 patients (5.1%) of the BVS group and 34 patients (4.2%) of the EES group (absolute difference 0.9%, 95% confidence interval [CI]: −1.2%-3.0%, p non-inferiority <0.001). Definite or probable device thrombosis (2.0% vs 0.6%, hazard ratio [HR] 3.32, 95% CI: 1.22-8.99, p=0.012) and TVMI (4.0% vs 2.1%, HR 1.96, 95% CI: 1.10-3.51, p=0.02) were significantly higher in the BVS group than in the EES group.
Conclusions: In patients at high risk of restenosis, non-inferiority of BVS compared with EES in terms of TLF was met at one year. BVS carried a higher risk of device thrombosis and TVMI than EES.
Introduction
The bioresorbable vascular scaffold (BVS) is designed for treatment of obstructive coronary artery disease providing temporary mechanical support and antiproliferative drug delivery, but without perceived disadvantages of permanent metallic implants1. In a series of randomised trials, BVS met the criteria of non-inferiority compared with metallic drug-eluting stents (DES) for composite endpoints in relatively low-risk coronary lesions and patients at one year2,3. However, BVS resulted in higher rates of target lesion failure (TLF) and device thrombosis compared with metallic DES at three-year follow-up4. These disappointing outcomes have been shown to be attributable, at least partially, to a suboptimal implantation technique of this thick-strut device5.
In previous randomised trials, the “BVS-specific” implantation technique was neither fully developed nor employed as part of the study design; whether using optimal implantation techniques with BVS could reduce the risk of device thrombosis requires further examination. In the DES era, prevention of in-stent restenosis and neoatherosclerosis remains an unmet need, especially for patients at high risk of restenosis, such as those with long lesions and patients with diabetes mellitus6. We hypothesised that the use of BVS in a high-risk population might demonstrate better long-term outcomes compared with DES after full BVS resorption. Therefore, we conducted the COMPARE-ABSORB trial to investigate the concept of short-term equivalence and long-term benefit of BVS over metallic DES in patients at high risk of restenosis with complex lesion(s).
Methods
The study design has been published previously7. In summary, the COMPARE-ABSORB trial is a prospective, randomised, controlled, single-blind, multicentre study across 45 centres in Europe (Supplementary Table 1). Patients aged 18-75 years with symptomatic ischaemic heart disease and presence of high-risk features for restenosis due to clinical profile or coronary lesion complexity and who were scheduled to undergo elective or emergent percutaneous coronary intervention (PCI) were eligible. Subjects participating in the trial met at least one of the inclusion criteria: medically treated diabetes, or multivessel disease with more than one de novo target lesion and/or presence of at least one complex target lesion (long lesion, small vessel, total occlusion or bifurcation). Key exclusion criteria included target lesion not suitable for BVS implantation, patients with cardiogenic shock, severe renal failure, severe impaired ejection fraction, left main disease or being on oral anticoagulants. Detailed criteria are listed in Supplementary Table 2. Patients were 1:1 randomly assigned to receive either BVS (Absorb™; Abbott Vascular, Santa Clara, CA, USA) or EES (XIENCE; Abbott Vascular). Block randomisation was performed with randomly selected block sizes. A dedicated implantation technique was defined in the protocol: predilatation using non-compliant balloons of the same diameter as the reference vessel diameter (RVD) and post-scaffold high-pressure (≥16 atm) dilatation were mandatory in the BVS group. Scaffold to vessel sizing was based on the instructions for use. The primary endpoint was TLF (a composite of cardiac death, myocardial infarction in the target vessel territory and clinically indicated target lesion revascularisation) at one year. An extended Methods section is provided in Supplementary Appendix 1, including study organisations (Supplementary Table 3), study procedures, hypotheses, endpoints (Supplementary Table 4), five definitions of optimal implantation technique (OIT) (Supplementary Table 5), and protocol revisions. Follow-up is planned for all patients up to seven years. Consideration will be given to extending follow-up to 10 years.
STATISTICAL ANALYSIS
All clinical data were analysed according to the intention-to-treat principle. For time-to-event endpoints, Kaplan-Meier plots were constructed and compared using the log-rank test. Binomial variables were evaluated with Fisher’s exact probability test, continuous variables tested with a two-sample t-test or with the Mann-Whitney U test when data were not normally distributed, and a p-value for interaction was calculated for the subgroup analyses.
The device implantation was evaluated in a combination of different parameters of predilatation, central core lab quantitative coronary angiography (QCA) sizing and post-dilatation. A two-sided p-value of less than 0.05 was considered to indicate statistical significance. All statistical analyses were performed using SAS software, Version 9.3 (SAS Institute Inc., Cary, NC, USA). This trial was registered at ClinicalTrials.gov (NCT 02486068).
Results
BASELINE PATIENT CHARACTERISTICS AND FOLLOW-UP
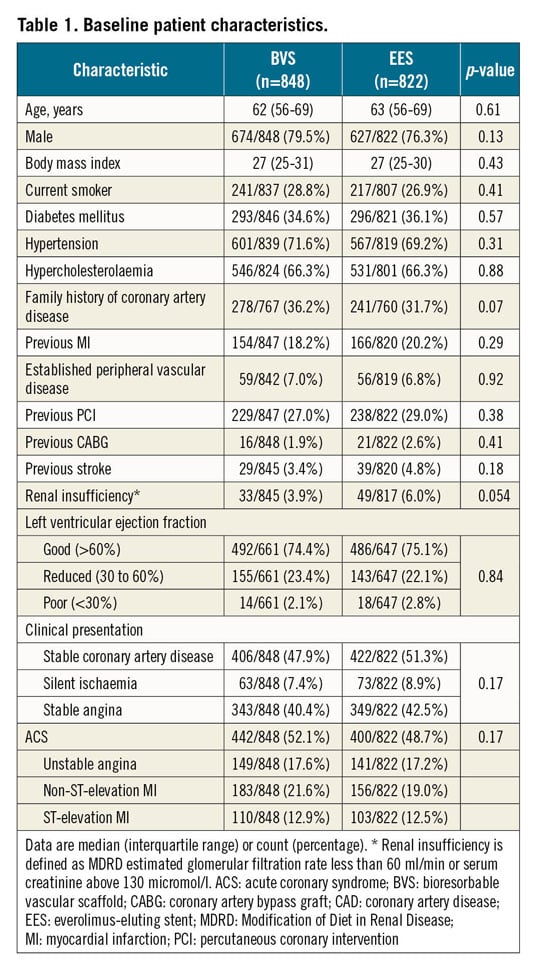
Between September 2015 and August 2017, 1,670 of the intended 2,100 patients with 2,457 lesions were randomly assigned to receive either BVS (848 patients with 1,243 lesions) or EES (822 patients with 1,214 lesions). The trial was prematurely stopped on the recommendation of the Data and Safety Monitoring Board based on safety concerns seen in interim analyses. Follow-up at 12 months was complete in 824/848 patients treated with BVS versus 804/822 patients in the EES group (p=0.43). In the 24 BVS patients with incomplete follow-up, the median duration of follow-up was 74 days versus 160 days in the 18 EES patients (p=0.36) (Figure 1). Baseline clinical characteristics are shown in Table 1. Of the 1,670 patients, 293 (34.6%) in the BVS group and 296 (36.1%) in the EES group had a history of diabetes, and 442 (52.1%) in the BVS group and 400 (48.7%) in the EES group presented with acute coronary syndrome.
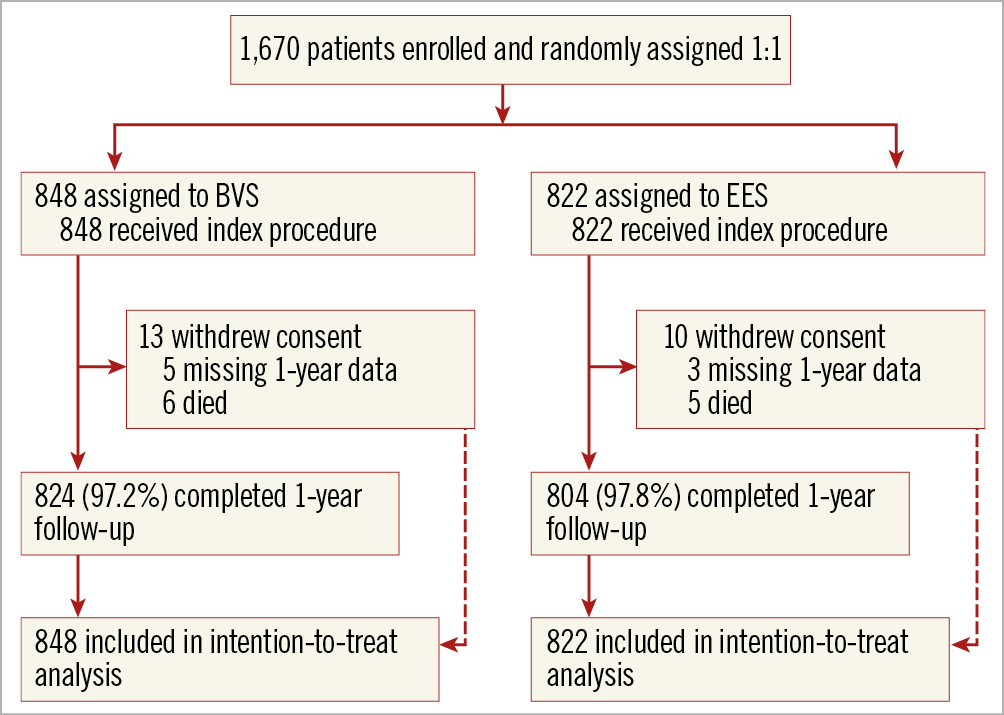
Figure 1. Study flow chart. BVS: bioresorbable vascular scaffold; EES: everolimus-eluting stent
LESION AND PROCEDURAL CHARACTERISTICS
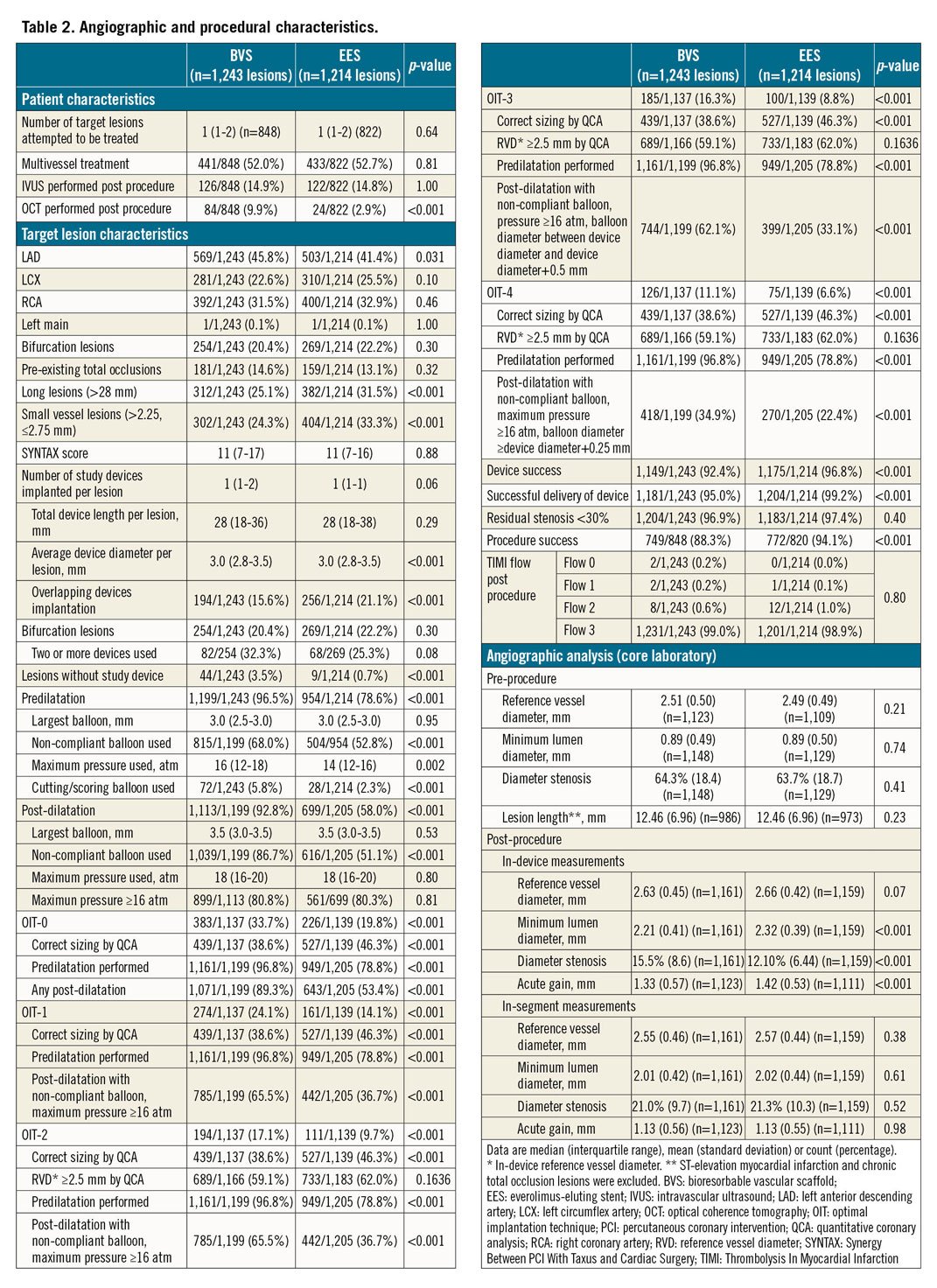
A total of 1,650 BVS and 1,554 EES were implanted in the two groups. Table 2 shows a significantly higher performance of optical coherence tomography (OCT) in the BVS group, as well as higher incidences of both long lesions (>28 mm) and small-vessel lesions (between 2.25 and 2.75 mm) in the EES group. In the BVS group, predilatation was performed in 96.8% of the lesions and post-dilatation in 92.8% of the lesions. The percentages of predilatation and post-dilatation were significantly higher in the BVS group than in the EES group. According to angiographic analysis performed by the core lab, 21.9% (255/1,166) of lesions in the BVS group had a post-procedural in-scaffold RVD of less than 2.25 mm and 40.9% (477/1,166) less than 2.5 mm. The main failures with respect to the sizing recommendation were related to differences between the visually estimated diameters and QCA, followed by mismatch between the proximal and distal reference diameters. The proportions of lesions in the BVS group meeting the definition of OIT-0 to OIT-4 were as follows: OIT-0: 33.7% (383/1,137), OIT-1: 24.1% (274/1,137), OIT-2: 17.1% (194/1,137), OIT-3: 16.3% (185/1,137), and OIT-4: 11.1% (126/1,137). In addition, the device success rate was significantly lower in the BVS group than in the EES group (BVS 92.4% vs EES 96.8%, p<0.001), driven by a lower rate of successful device delivery.
CLINICAL OUTCOMES
The primary endpoint of TLF at one year occurred in 43 patients (5.1%) in the BVS group and in 34 patients (4.2%) in the EES group (Table 3, Figure 2). The primary hypothesis, non-inferiority of BVS compared to EES, was met with an absolute difference of 0.9% and two-sided 95% confidence interval (CI) of −1.2% demonstrated (difference 0.9% [two-sided 95% CI: −1.2-3.0%], p non-inferiority <0.001). For individual components of TLF, the frequencies of cardiac death (5 [0.6%] vs 1 [0.1%]; hazard ratio [HR] 4.87, 95% CI: 0.57-41.7; p=0.11) and clinically indicated target lesion revascularisation (20 [2.4%] vs 22 [2.7%]; HR 0.89, 95% CI: 0.48-1.62; p=0.69) did not differ significantly between the groups. However, at 30 days, target vessel myocardial infarction (TVMI) was significantly higher in the BVS group than in the EES group (27 [3.2%] vs 10 [1.2%]; HR 2.64, 95% CI: 1.28-5.45; p=0.006), while no significant difference between groups beyond 30 days to one year (7 [0.9%] vs 7 [0.9%]; HR 0.99, 95% CI: 0.35-2.83; p=0.99) was observed (Table 3, Figure 3). Similarly, definite or probable device thrombosis was significantly higher in the BVS group than in the EES group (13 [1.5%] vs 4 [0.5%]; HR 3.16, 95% CI: 1.03-9.69; p=0.033) at 30 days and was not significantly different between 30 days and one year (4 [0.5%] vs 1 [0.1%]; HR 3.94, 95% CI: 0.44-35.2; p=0.18). The majority of the BVS thromboses (13/17, 76%) occurred within 30 days of the index procedure and only one event was related to the cessation of antiplatelet agents.
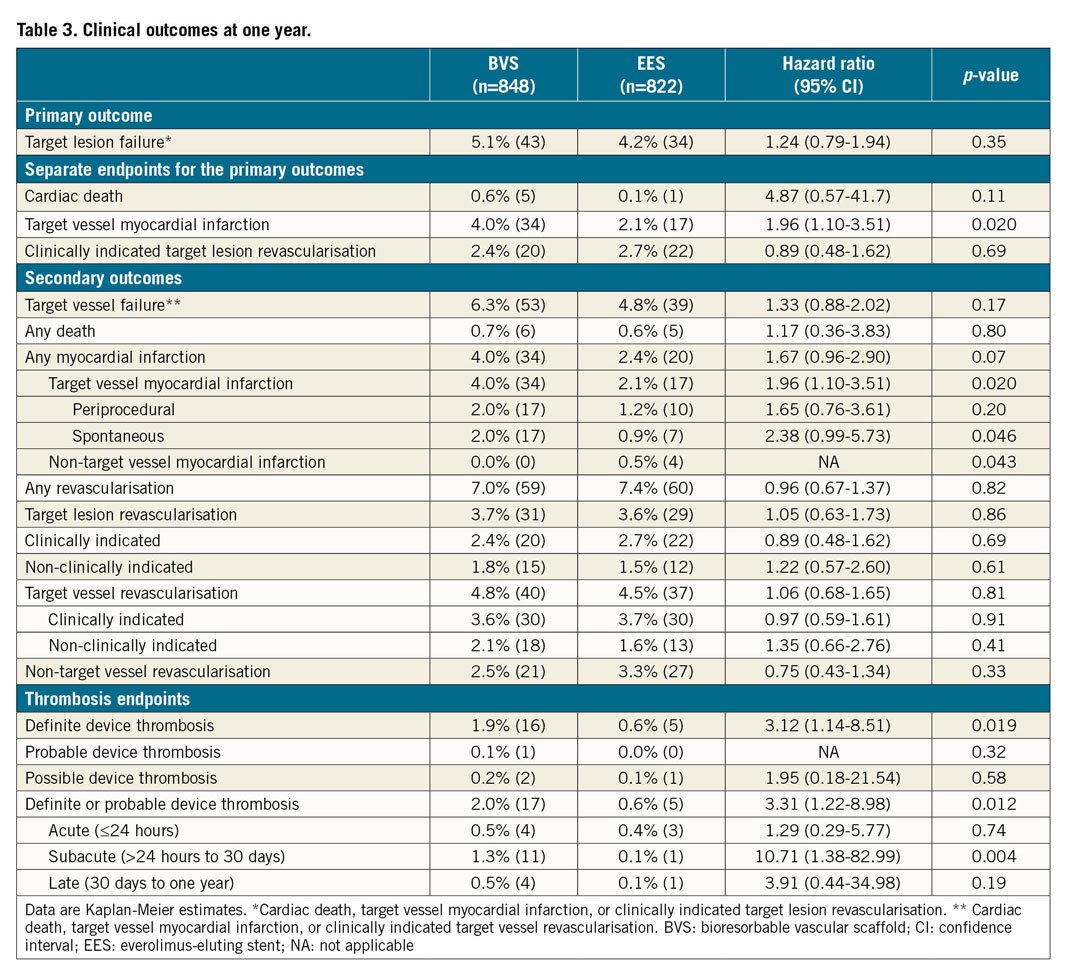
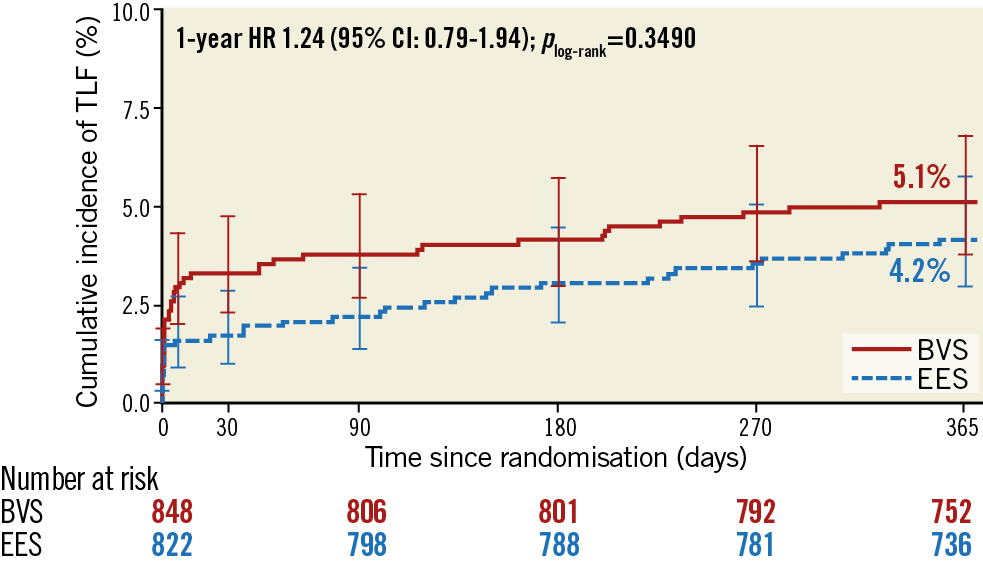
Figure 2. Kaplan-Meier plot for the primary endpoint. Kaplan-Meier curves show the cumulative incidence of target lesion failure. BVS: bioresorbable vascular scaffold; CI: confidence interval; EES: everolimus-eluting stent; HR: hazard ratio; TLF: target lesion failure
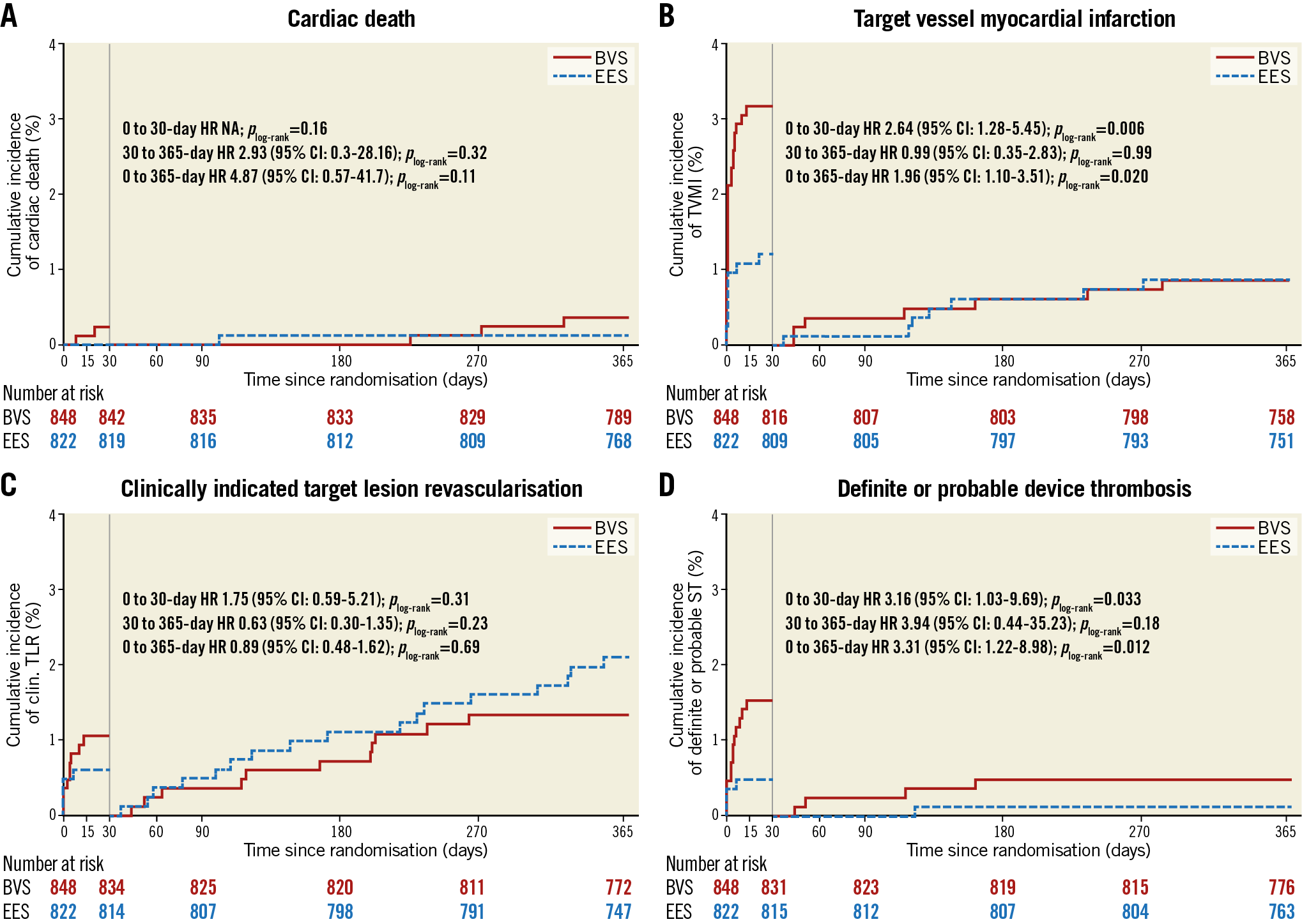
Figure 3. Kaplan-Meier plots for the components of the primary endpoint and definite/probable device thrombosis. A) Cardiac death. B) Target vessel myocardial infarction. C) Clinically indicated target lesion revascularisation. D) Definite/probable device thrombosis. BVS: bioresorbable vascular scaffold; CI: confidence interval; EES: everolimus-eluting stent; HR: hazard ratio; NA: not applicable; ST: stent thrombosis; TLR: target lesion revascularisation; TVMI: target vessel myocardial infarction
At one-year follow-up, 80.0% of patients in the BVS group and 70.8% of patients in the EES group remained on dual antiplatelet treatment (Supplementary Table 6). The one-year TLF rates were comparable across all pre-specified subgroups (Figure 4).
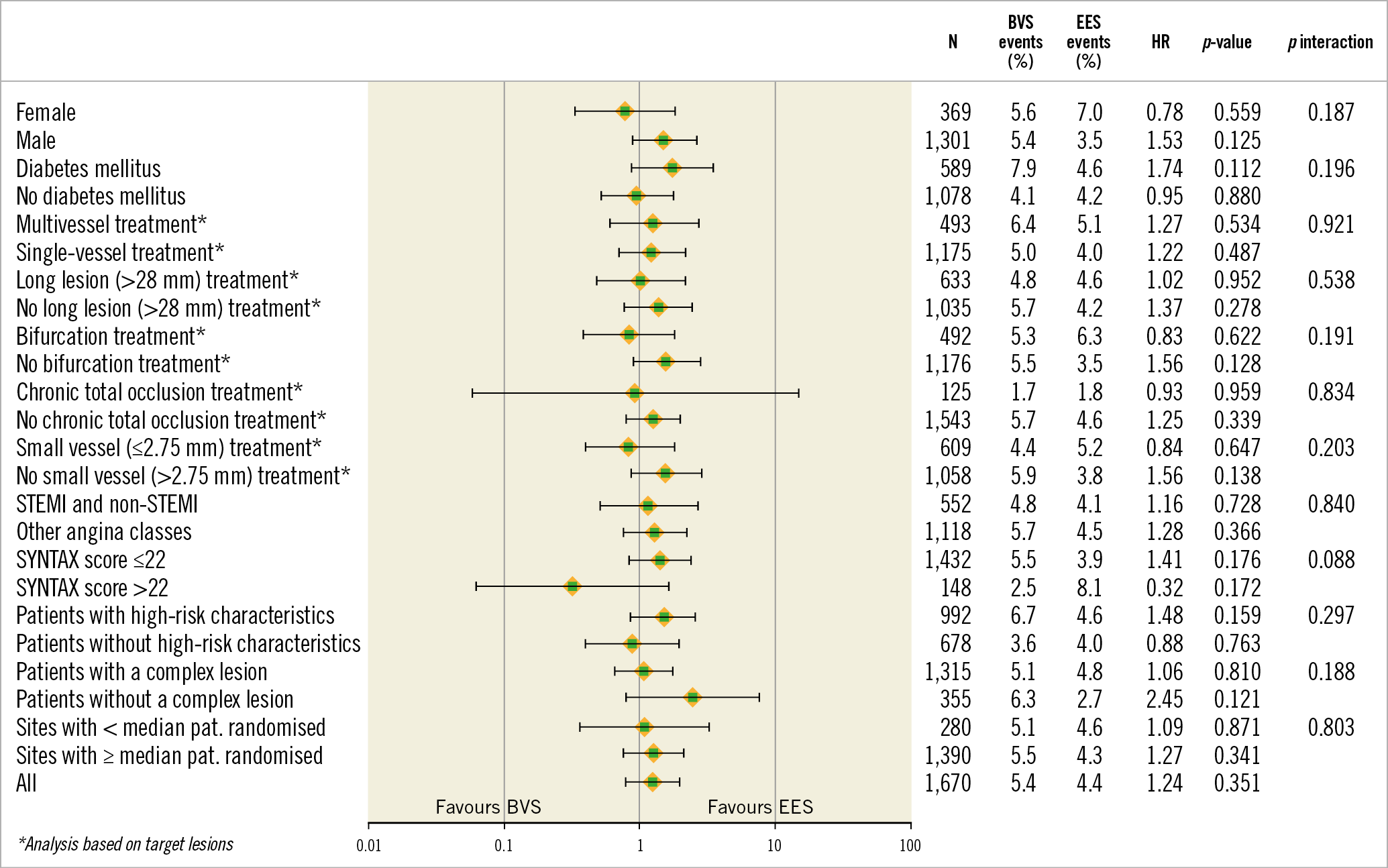
Figure 4. Stratified analyses of the primary endpoint across subgroups. Hazard ratio with 95% CI and p-value results were from Cox proportional hazards analysis. BVS: bioresorbable vascular scaffold; EES: everolimus-eluting stent; HR: hazard ratio; N: number of patients; STEMI: ST-elevation myocardial infarction
Additional post hoc analyses with respect to the TLF and definite and probable device thrombosis rates stratified by OIT did not show significant treatment-by-subgroup interactions (Supplementary Figure 1A, Supplementary Figure 1B). The definite device thrombosis rates did not differ significantly between lesions with or without a correct vessel sizing, though a combined proximal and distal oversizing of the scaffold showed a high event rate (Supplementary Figure 2). Results of quality of life reported at follow-up are shown in Supplementary Table 7.
Discussion
In the present large-scale, randomised trial, the BVS was non-inferior to the EES in terms of TLF at one year in a population at high risk of restenosis. Moreover, the treatment effect on TLF was similar across different subgroups, including risk groups defined according to lesion complexity or baseline characteristics. Although non-inferiority of the primary endpoint was met, definite or probable device thrombosis and TVMI rates were significantly higher in the BVS group compared with the EES group, which resulted in premature cessation of recruitment to the study.
Compared with the one-year results of the ABSORB III trial, the device thrombosis rate in the BVS group was slightly higher in the present study (2.0% vs 1.5%), whilst it was similar in the EES group (0.6% vs 0.7%). The observed higher device thrombosis rate in this trial is probably attributable to the complexity of patients and lesions included. Furthermore, device thrombosis events occurred predominantly during the early phase after implantation, implicating procedure-related causes. According to lessons learned from previous studies, a mismatch between vessel size and device size is a predictor of early and late scaffold thrombosis8. Furthermore, the meta-analysis on ABSORB trials incriminated BVS for vessels with a diameter of less than 2.5 mm9. Theoretically, these problems might be reduced by implementing an optimal implantation technique. The COMPARE-ABSORB study excluded vessels smaller than 2.25 mm in the original study design, then amended the exclusion criterion of minimal vessel size to 2.5 mm during enrolment because of these safety concerns. Investigators were also advised to estimate the vessel size by quantitative coronary analysis or intravascular imaging if the vessel size was less than 2.75 mm by visual assessment. Nevertheless, the post hoc angiographic analysis performed by the core lab showed that 40.9% of lesions in the BVS group had a post-procedural RVD smaller than 2.5 mm. These findings emphasise the importance of appropriate vessel sizing, which could not be truly achieved by visual assessment alone. Because underestimation of vessel size by QCA is a limitation of angiography10, mandatory intravascular imaging guidance should be explored in future when implanting BVS in order to enhance safety. On the other hand, only a minority of lesions (33.7%) fit the OIT criteria due to sizing mismatch (the lesion segment not fitting any scaffold diameter due to incompatibility of proximal and distal reference requirements in sizing for a specific scaffold diameter). In the BVS group, 11.5% of lesions had both proximal and distal reference diameters within the range of correct sizing, which was defined as device size ± 0.25 mm, 27.1% had either proximal or distal reference diameter within the range but not both, and 61.4% had both reference diameters out of the range (Supplementary Table 8). This demonstrates that correct sizing with BVS according to the OIT criteria is difficult to achieve in the majority of lesions with one BVS. Further improvements to the device such as thinner and smaller struts, better conformability and radial strength are therefore indispensable.
In the COMPARE-ABSORB trial, high-pressure post-dilatation with a non-compliant balloon was mandated by the protocol. Nevertheless, based on angiographic analysis, acute gain and established post-procedural minimal lumen diameter in the BVS arm did not match those of the EES arm, although the absolute differences between arms appeared to be smaller than or similar to the differences observed in previous trials (Supplementary Table 9). This unclosed gap in acute performance between the devices could be a contributory factor for early scaffold thrombosis with BVS compared with EES.
Recently, the five-year results of the ABSORB II trial showed a significant difference in TLF in favour of EES compared to BVS (Serruys PW. The 5-year Clinical Outcomes of the ABSORB II Trial: First Randomized Comparison between the Absorb Everolimus Eluting Bioresorbable Vascular Scaffold and the XIENCE Everolimus Eluting Stent. Presented at Transcatheter Cardiovascular Therapeutics, San Diego, CA, USA, 22 September 2018). This significant difference in TLF was driven by events that had occurred within the first three years. No scaffold thrombosis was observed between three and five years. Therefore, extension of follow-up duration from five to seven years in this study is necessary to determine whether more normalised coronary function and physiology after complete scaffold bioresorption will provide a clinical advantage for BVS over metallic DES.
Limitations
First of all, despite the fact that optimal implantation technique was incorporated in the study design, on-line QCA or intravascular imaging was suggested, but not mandatory, for vessel sizing. Secondly, the one-year TLF rates for both devices were remarkably lower than anticipated and therefore the non-inferiority margin of 4.5% was relatively wide. However, the non-inferiority margin was in line with the ABSORB III study, in which the U.S. Food and Drug Administration was consulted. In the study design of ABSORB III11, the assumed rate of the primary endpoint was 7.0%. The ratio of non-inferiority margin to assumed event rate was 64%. In our study, the assumed event rate was 8.5% and the ratio of non-inferiority margin to assumed event rate was 53%, which was slightly stricter than that in ABSORB III. Thirdly, because of significant differences in predilatation and post-dilatation rates between the stent arms, we cannot exclude an influence on outcomes caused solely by differences in implantation technique. Fourthly, bleeding event was not a pre-specified endpoint and thus not reported in this paper. Lastly, the study results apply only to the BVS, which is no longer commercially available for use in clinical practice. Nevertheless, the COMPARE-ABSORB study was the first trial to investigate the performance of BVS in complex lesions and high-risk patients.
Conclusions
In the present large-scale randomised trial of patients at high risk of restenosis, BVS was non-inferior to EES for the primary endpoint, TLF at one year. BVS carried a higher risk for device thrombosis and TVMI, especially in the early stages after implantation.
|
Impact on daily practice This trial showed that using an optimal implantation technique did not prevent an increase of device thrombosis with BVS at one year. Further exploration of the long-term benefit after BVS implantation and device modification is warranted. |
Acknowledgements
All authors are indebted to Prof. Patrick W. Serruys (Imperial College London, London, UK) as a senior consultant to the trial and to Mrs Ute Windhovel (CERC, Massy, France) and Mrs Ria van Vliet (Maasstad CardioVascular Research, Rotterdam, the Netherlands) for assistance in coordinating the trial.
Funding
The trial sponsor was Maasstad Hospital, Rotterdam, the Netherlands, which received an institutional grant from Abbott Vascular. The grant giver was not involved in the conduct, data management, or data analysis of the trial, conducted by CERIC, Geneva, Switzerland, which contracted CERC and Cardialysis as clinical research organisation (CRO) and core lab.
Conflict of interest statement
P.C. Smits has received institutional research grants and speaker fees from Abbott Vascular, St. Jude Medical and Terumo. B. Chevalier has received grants and personal fees from Abbott Vascular during the conduct of the study, and personal fees from Medtronic, Terumo, and Biotronik, and is a shareholder and general director of CERC (CRO), outside the submitted work. N. West has received speaker fees from Abbott Vascular. T. Gori has received speaker fees from Abbott Vascular. E. Barbato has received personal fees from Boston Scientific, Abbott Vascular, Opsens Medical, and GE Healthcare, outside the submitted work. V. Kočka has received personal fees from Abbott Vascular, Medtronic, B Braun, and Terumo, outside the submitted work. J. Tijssen has received grants and personal fees from Abbott Vascular during the conduct of the study. M.C. Morice is the CEO of CERC, the CRO which conducted the trial. Y. Onuma was an advisory board member of Abbott Vascular. R.J. van Geuns reports grants and personal fees from Abbott Vascular, outside the submitted work. G. Tarantini reports personal fees from St. Jude Medical, personal fees from Edwards Lifesciences, Boston Scientific, Abbott Vascular, and AstraZeneca, outside the submitted work. J. Escaned reports consultancies and/or speaker fees at educational events for Abbott, Biotronik, Boston Scientific, Medtronic, Opsens, OrbusNeich, and Philips Healthcare. The other authors have no conflicts of interest to declare.
Supplementary data
To read the full content of this article, please download the PDF.
