
Abstract
Aims: The novel sirolimus-eluting ultra-high molecular weight APTITUDE bioreabsorbable vascular scaffold (BRS) displays higher mechanical strength, expansion capabilities and resistance to fracture compared to other BRS technologies. RENASCENT II is a prospective, multicentre first-in-human clinical study evaluating the clinical performance of the APTITUDE BRS in the treatment of single de novo coronary lesions among patients undergoing percutaneous coronary intervention.
Methods and results: The APTITUDE BRS was tested in a prospective study in two countries (Italy and Colombia). Study objectives were angiographic in-scaffold late lumen loss (IS-LLL) measured by quantitative coronary angiography (QCA) and target vessel failure (TVF) defined as the composite rate of cardiac death, target vessel myocardial infarction (TV-MI) or ischaemia-driven target lesion revascularisation (TLR) at 9 and 24 months. A total of 60 patients were enrolled. All patients underwent lesion predilatation and 46 patients (76.7%) underwent post-dilatation. Clinical device and procedural success were 98.3% (59/60 patients) and 100%, respectively. Angiographic late lumen loss was 0.19±0.26 mm at 9 months and 0.3±0.41 mm at 24 months. At 9 months, TVF occurred in 2/59 patients (3.4%) due to TV-MI but there was no TLR. No further cases of TVF, MACE or stent thrombosis were reported up to 24-month follow-up.
Conclusions: In this multicentre prospective study, the APTITUDE BRS was shown to be safe and effective in the treatment of single coronary lesions at 24-month clinical follow-up.
Introduction
Current drug-eluting stents (DES) are safe and have very low thrombosis rates1. However, the potential limitations of DES include the permanent presence of a metallic foreign body within the artery and often a durable polymer, either of which may cause vascular inflammation, neoatherosclerosis and restenosis or perpetuate the risk of very late stent thrombosis2. Moreover, metallic stents indefinitely impair the physiological vasomotor function of the vessel and also the potential for future grafting within the stented segment3,4. In this context, bioresorbable scaffolds (BRS) represent the latest innovation in the field of percutaneous coronary intervention (PCI). They aim to provide a transient vessel scaffold, preventing acute vessel closure/recoil and subsequently dissolve. In addition, complete bioresorption of the scaffold is associated with plaque regression, late vessel lumen enlargement and restoration of vasomotion within a few years. Thus, BRS hold the potential to achieve the paradigm of vascular restoration therapy, restoring both vessel lumen and vascular function eliminating the risk of late stent-related events.
However, BRS have several limitations including thicker, wider struts, less radial strength and limited expansion capabilities. These limitations require altered implantation techniques to those of standard DES, especially in complex coronary artery disease. To counteract the lower radial strength ascribable to the nature of their manufacturing, some companies have designed their BRS products with struts thicker than most second-generation DES. Furthermore, BRS have been shown to have an increased stent thrombosis risk compared to metallic DES, particularly very late stent thrombosis (VLST)5,6. Scaffold dismantling related to scaffold reabsorption was found to be the commonest mechanism of VLST in the INVEST registry7.
New-generation thinner BRS implanted using an optimal technique might offer early and intermediate-term outcomes comparable to contemporary metallic DES (prior to complete bioresorption), with improved long-term event-free survival.
The reduction of strut thickness from the current 150 μm BRS to the newer-generation scaffolds having 100-120 μm struts may reduce flow disturbances and hence thrombogenicity8. There are multiple newer-generation BRS at different stages of development with varying mechanical or bioresorption properties. RENASCENT III is a first-in-man (FIM) clinical safety trial of the newest (98 μm strut thickness) BRS MAGNITUDE® (Amaranth Medical Inc., Mountain View, CA, USA). These new BRS first have to show clinical safety in FIM trials and subsequently be tested further in randomised controlled trials (RCT) with proven metallic DES.
The aim of the RENASCENT II trial is to evaluate the clinical and safety performance of the APTITUDE® (Amaranth Medical Inc.) BRS.
Methods
STUDY DESIGN AND PATIENT POPULATION
The RENASCENT II study is a prospective, non-randomised, non-inferiority study of the APTITUDE bioresorbable drug-eluting coronary scaffold (NCT02568462) that enrolled 60 patients from Colombia and Italy. The ethics committee at each participating institution approved the protocol and each patient gave written informed consent before inclusion. As required by national regulations, the approval of the relevant national regulatory agency was also obtained.
Inclusion and exclusion criteria are shown in Supplementary Table 1.
STUDY DEVICE
The APTITUDE design is based on the FORTITUDE® scaffold (Amaranth Medical Inc.). The FORTITUDE scaffold has been demonstrated to be biocompatible and to maintain mechanical integrity with controlled drug release in previous trials. The key design difference between the two is a reduction of strut thickness (APTITUDE 115 μm vs FORTITUDE 150 μm). The scaffold material (ultra-high poly-L-lactic acid [PLLA]), manufacturing process and delivery system have not changed.
The APTITUDE BRS is made with a continuous “closed cell” zigzag helical design made of ultra-high PLLA and coated with a polymer-antiproliferative drug matrix (poly-L-lactic acid + sirolimus) mixed in a 1:1 polymer to drug ratio with 90% of the drug being released by 90 days.
In vitro studies have shown that the scaffold degrades over time with the reduction in molecular weight reaching approximately 50% at 8 months and greater than 85% at 18 months. The radial support is maintained for 8 to 10 months9. As the scaffold degrades, the polymer is converted into lactic acid, which is metabolised through the Krebs cycle. The degradation process takes approximately two years and is very similar to that of the Abbott BVS bioresorbable scaffold.
The polymer and design of the scaffold provide uniform strength in all directions. This uniform strength also makes the scaffold less likely to fracture or crack in high stress areas.
Supplementary Table 2 shows features of the APTITUDE BRS. Figure 1 shows optical coherence tomography (OCT) images comparing the Abbott BVS, and the Amaranth FORTITUDE and APTITUDE.
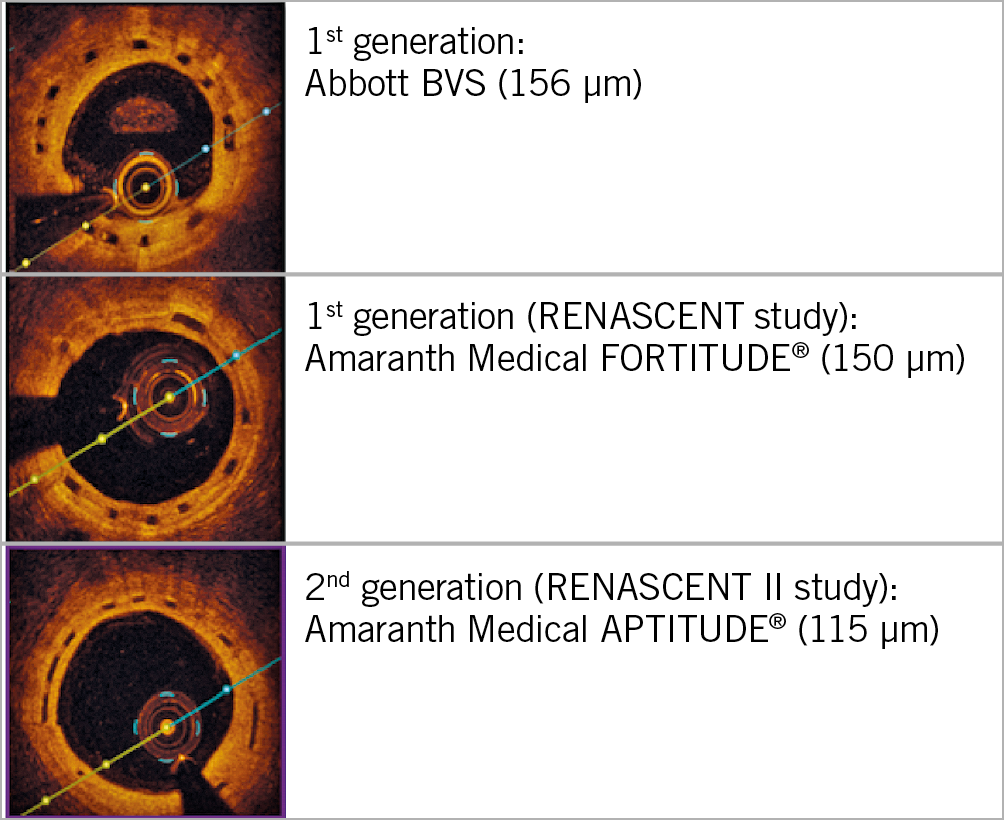
Figure 1. OCT images showing the APTITUDE BRS with thinner struts (9 months post implantation) in comparison with the Abbott BVS and FORTITUDE BRS.
STUDY PROCEDURE
Target lesions were treated using standard interventional techniques; successful predilatation of the target lesion was mandatory (1:1). Baseline intravascular ultrasound (IVUS) assessment was performed during the index procedure to evaluate vessel size and degree of calcification, and to determine the appropriate scaffold size. The target lesion had to be treated with a single study device and planned overlapping with another stent was not allowed. Post-dilatation was not mandatory but allowed at the operator’s discretion (if the angiographic result was suboptimal) using a non-compliant balloon with diameter ≤0.5 mm larger than the nominal scaffold size. Bail-out stenting with DES for non-flow-limiting edge dissection was recommended and, as per clinical practice, required for flow-limiting dissection. Post-procedural intravascular imaging with OCT was required in all cases.
Treatment with aspirin was started at least 24 hours before the procedure and a ≥75 mg/day dose was required for the duration of the study. A loading dose of ≥300 mg clopidogrel (or 60 mg prasugrel/180 mg ticagrelor) was administered before the procedure, followed by 75 mg clopidogrel daily (or 10 mg prasugrel daily/90 mg ticagrelor twice daily) for a minimum of 12 months. The duration of dual antiplatelet therapy beyond 12 months was left to the discretion of the physician.
The 30-day follow-up was performed via an office visit or by phone call. At nine months, angiographic follow-up with OCT was performed. Coronary computed tomography angiography (CTA) or invasive coronary angiography was carried out at 24 months, depending on centre preference. Colombian centres performed invasive coronary angiography while Italian centres preferred to use coronary CT. All data were collected in dedicated electronic case report forms. The study stopped at the end of 24 months.
STUDY OBJECTIVES
The primary performance endpoint was in-scaffold late lumen loss (IS-LLL), defined as the amount of vessel lumen diameter lost/gained at the time of angiographic follow-up measured by quantitative coronary angiography (QCA) at nine months. The assessment was made within the segment of vessel including the scaffold.
The primary safety endpoint was the incidence of target vessel failure (TVF), defined as cardiac death (Academic Research Consortium [ARC] definition)4, target vessel myocardial infarction (TV-MI) (using the expert consensus document from the Society for Cardiovascular Angiography and Interventions [SCAI])10, or clinically indicated target lesion revascularisation (TLR) (ARC definition) at nine months. Although the adjudication of periprocedural MI was performed using the SCAI definition, additional analyses were performed using the third universal definition of MI6. Stent thrombosis was defined using the ARC “definite” or “probable” stent thrombosis definitions4.
Furthermore, both “clinical device success”, defined as successful delivery and deployment of the clinical investigation scaffold with a final residual stenosis of <50% by QCA after the index procedure, and “clinical procedure success”, defined as clinical device success using any adjunctive device without occurrence of major adverse clinical events related to ischaemia up to day of discharge, were assessed.
Meditrial Europe Ltd (Zürich, Switzerland) was responsible for the submission of the protocol to the relevant ethics committees and authorities, monitoring of the patients’ data and the reporting of serious adverse events to the respective authorities for the RENASCENT II trial. Adverse events were adjudicated by an independent clinical events committee. An independent core lab (Cardiovascular Research Foundation, New York, NY, USA) performed angiographic (QCA), OCT and CTA data analysis.
Angiographic, QCA, OCT image acquisition and data analysis are described in Supplementary Appendix 1-Supplementary Appendix 4, Supplementary Figure 1 and Supplementary Figure 2.
STATISTICAL ANALYSIS
Angiographic IS-LLL at nine months was analysed using a one-sample t-test for non-inferiority. If the assumptions for normality were not met, then a Wilcoxon signed-rank test was used. When provided, the 95% confidence intervals were computed with the Gaussian approximation, taking into account the paired analysis. Paired comparisons between post-procedural and follow-up results were performed using a Wilcoxon signed-rank test. The results for the endpoints are presented using summary statistics and 95% confidence intervals. For discrete outcomes, the total number and percentage are presented.
Results
BASELINE CLINICAL AND ANGIOGRAPHIC CHARACTERISTICS
The baseline clinical and angiographic characteristics of the study population are shown in Table 1. A total of 60 patients were enrolled, 23 in Colombia and 37 in Italy. The mean reference vessel diameter was 2.8±0.4 mm and lesion length 12.4±3.6 mm. Most of the lesions were type ACC/AHA B1-C (83.3%, n=50). There was moderate-severe calcification in six cases (10%). Figure 2 shows the RENASCENT II study flow chart.
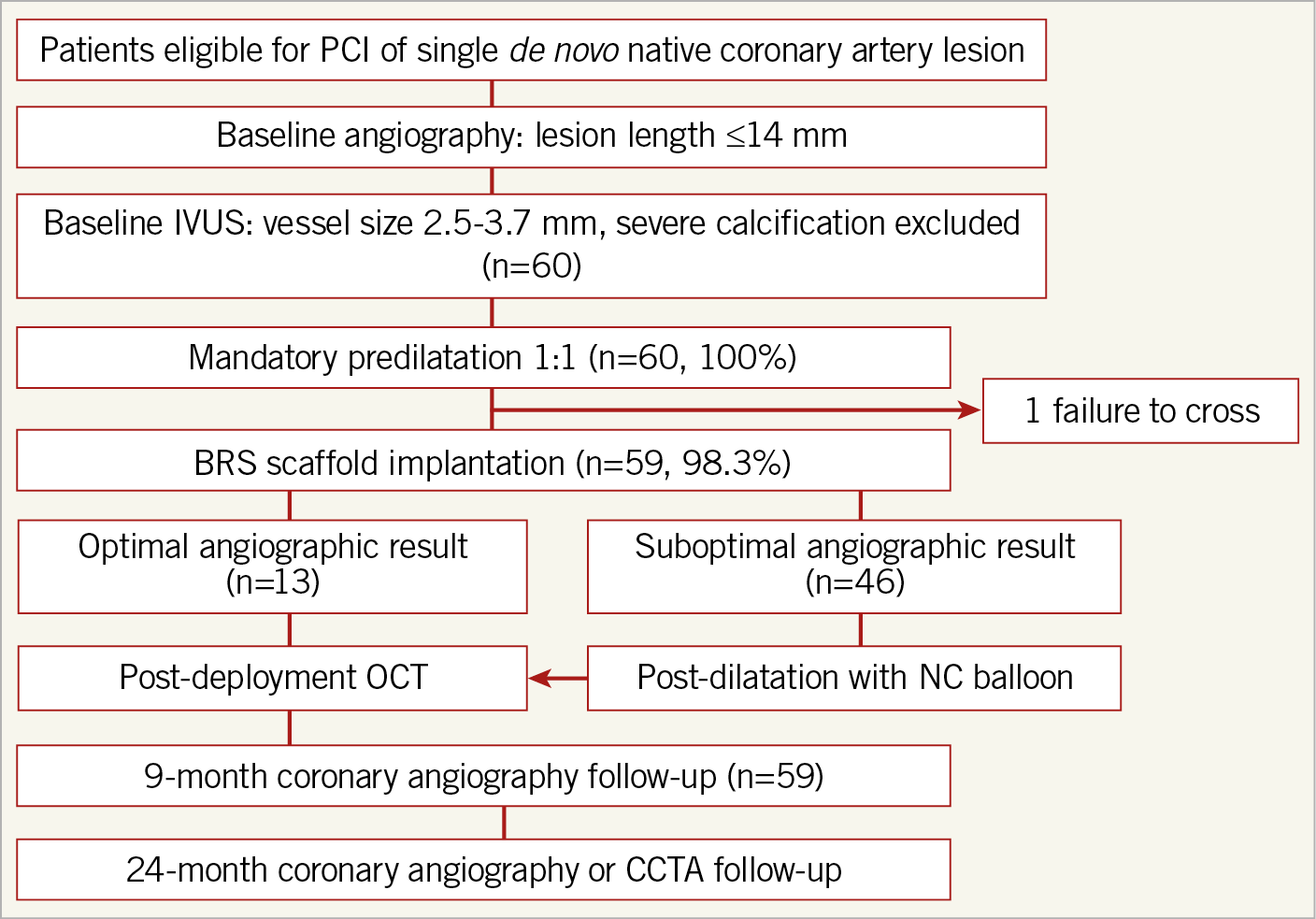
Figure 2. A flow chart of the APTITUDE study design.
PROCEDURAL CHARACTERISTICS
Table 2 shows the procedural characteristics of the study population. Baseline IVUS assessment was performed during the index procedure in all patients to evaluate vessel size and grading of calcification, and to select the appropriate scaffold size. Appropriate predilatation was performed in 100% of the lesions. In 76.7% (n=46) of cases, post-dilatation was performed. There were no dissections requiring a bail-out DES.
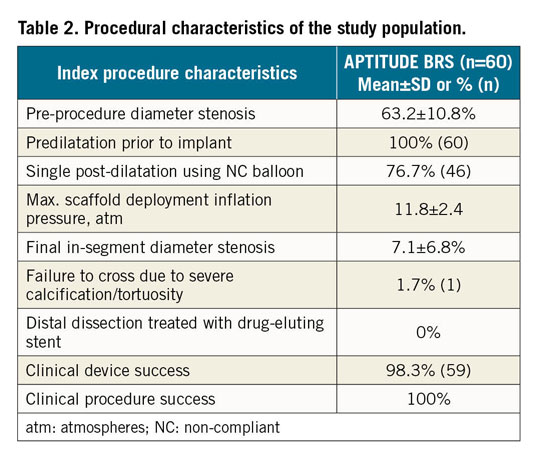
Clinical device success was 98.3% (n=59); in one case the scaffold was not implanted due to inability to track through a calcified and tortuous vessel proximal to the target lesion. The resulting clinical procedure success rate was 100% (n=60).
STUDY OBJECTIVES
Table 3 shows results of major adverse cardiovascular events (MACE) during the trial up to 24-month follow-up. There were no major cardiovascular events in hospital or up to 30-day follow-up.
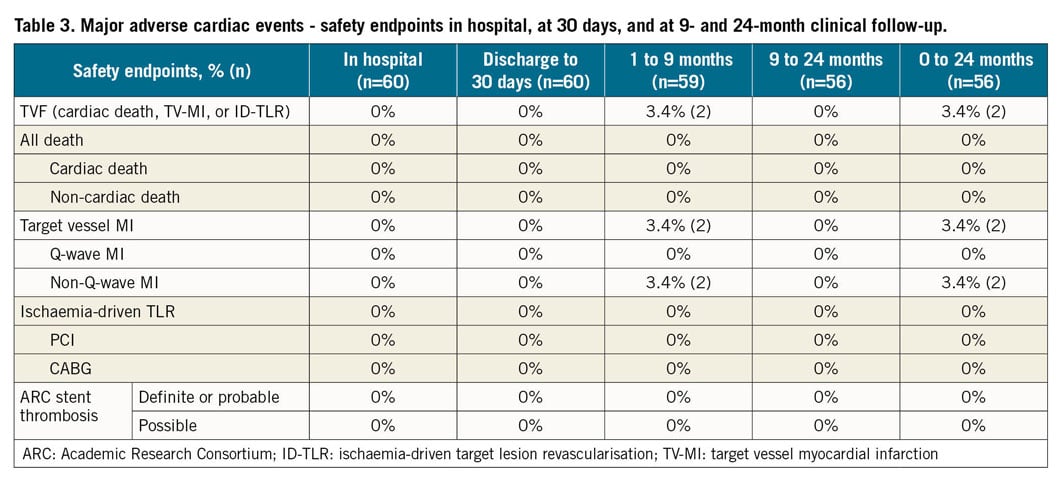
At nine months, 59 (98%) patients had completed clinical and mandatory angiographic follow-up. One patient did not receive the study device and, per protocol, exited the study at 30 days. At nine months, TVF was 3.4% (n=2) due to two non-Q-wave MIs (target vessel MIs) but there was no TLR. Details of these two cases are provided in Supplementary Table 3. No ischaemia-driven TLR or scaffold thrombosis was reported up to 24-month follow-up. There were two cases of binary stenosis at 24-month follow-up (Supplementary Table 4). However, these patients were asymptomatic and no intervention was required as it was not clinically indicated. At 24-month follow-up, 24 out of 55 patients (43.6%) were still on dual antiplatelet therapy.
ANGIOGRAPHIC AND QCA ANALYSIS
Table 4 shows QCA measurements at baseline, post scaffold implantation, and at 9- and 24-month follow-up. IS-LLL was 0.35±0.33 mm at 9 months and 0.37±0.44 mm at 24 months (Figure 3). Other significant QCA measurements were in-segment minimal luminal diameter (MLD) 1.0±0.3 mm at baseline, and in-scaffold MLD 2.9±0.4 mm post BRS implantation, 2.5±0.4 mm at 9 months and 2.3±0.6 mm at 24 months. There was an acute gain of 1.9±0.4 mm post BRS insertion.
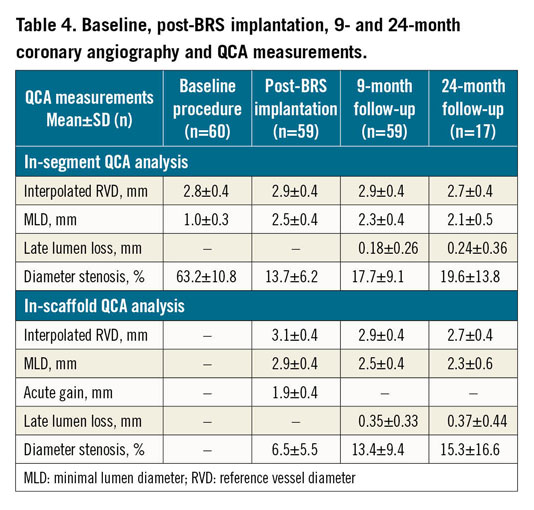
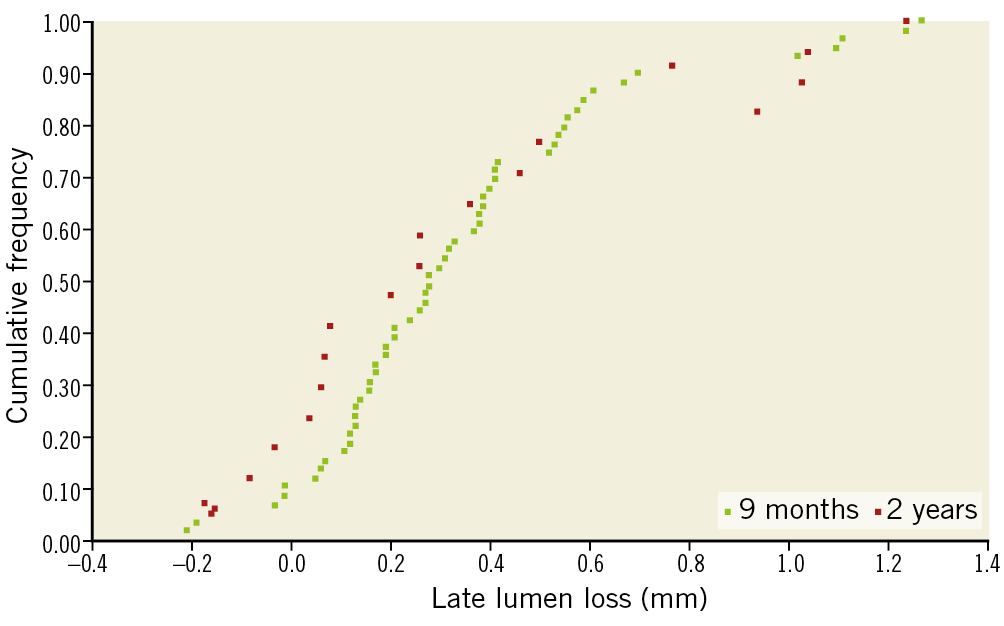
Figure 3. Cumulative frequency of in-scaffold late lumen loss at 9 and 24 months.
OCT ANALYSIS
OCT pullbacks were analysed in 53 lesions during the index procedure (post scaffold implantation) and 58 lesions at 9-month angiographic follow-up. Supplementary Table 5 shows the in-scaffold OCT measurements. The percentage of intra-scaffold neointimal hyperplasia (NIH) volume at 9 months was very low (13.3±6.1%). The total percentage of covered struts at 9 months was 97.0%, of which 96.52±5.02% were apposed to the vessel wall. The total percentage of uncovered struts at 9 months was very low (2.97%). Figure 4 shows the mean outer scaffold area in 51 matched patients at post implant and at nine months (for each patient).
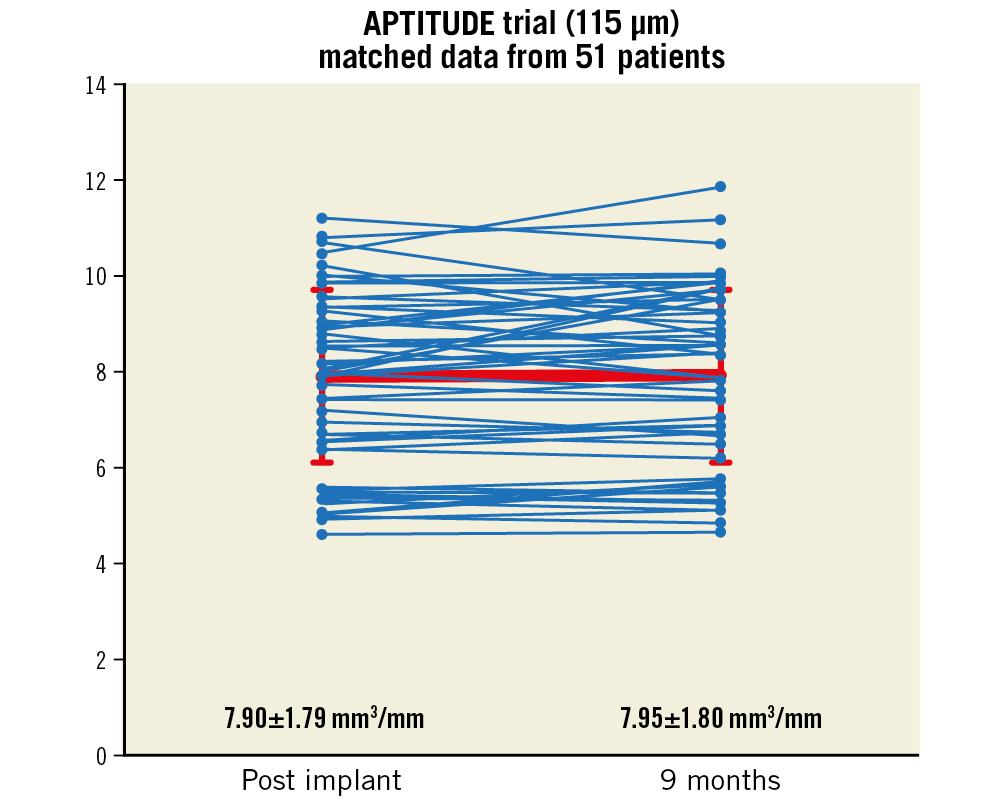
Figure 4. Scaffold integrity at 9 months: mean outer scaffold area by OCT for 51 patients post implant and at 9 months.
Discussion
The major findings of the international, multicentre study of the novel thin-walled 115 μm APTITUDE bioresorbable scaffold are the following: a) high clinical device success rate; b) low MACE rate up to 24-month follow-up (3.4%; both non-Q-wave MIs related to non-TLR) as expected in this population, c) no scaffold thrombosis, d) scaffold stability maintained up to 24 months, e) high level of strut coverage (97.0%) and low rate of malapposition (0.037%, all covered) as evidenced by OCT.
One of the major challenges in the BRS field has been the development of scaffolds displaying stent-like mechanical strength and resistance to the compressive load imposed by vessel recoil following deployment in challenging anatomical conditions9. In first-generation BRS, crystalline polymeric structures provided mechanical strength to the scaffold. However, the highly crystalline polymer structure limited the scaffold’s expansion capabilities and its resistance to fracture. As a result, current-generation BRS display limited expansion capabilities beyond pre-determined limits and are prone to fracture if not deployed properly.
The ultra-high molecular weight PLLA-based BRS have already displayed higher expansion capabilities and resistance to fracture under static and dynamic loading conditions9. At EuroPCR 2018, the RENASCENT trial showed good safety performance of the FORTITUDE BRS with lumen patency and vessel wall stability up to 24 months. This is once again reproduced in this trial with the low angiographic LLL rate (0.35 mm at 9 months and 0.37 mm at 24 months). Furthermore, OCT analysis showed high strut apposition and coverage rates at nine months.
ACUTE GAIN AND LATE LUMEN LOSS
In our analysis, the APTITUDE BRS showed an acute gain of 1.9±0.4 mm, the same as that reported for the FORTITUDE BRS. Ormiston et al reported an acute gain of 1.22±0.38 mm for the second generation of the Absorb™ BVS (Abbott Vascular, Santa Clara, CA, USA) in the ABSORB cohort B trial which was numerically lower compared to the EES (1.32±1.26 mm)6 . The numerically higher EES acute gain could be secondary to higher recoil rates or more conservative post-dilatation techniques used during BVS deployment aiming to avoid strut fractures5. The in vivo acute gain of the FORTITUDE BRS has been reported to be higher compared to the BVS. Cheng et al reported in vitro analysis that compared the capability of the Amaranth Medical BRS to resist fracturing under high load conditions9. They reported that the number of fractures was higher in BVS versus the FORTITUDE BRS with lower percentages of late scaffold recoil at three months.
Numerous studies have shown that LLL is a predictor of MACE. LLL provides an indirect angiographic evaluation of the vessel wall response to the metallic stent related to neointimal proliferation in metallic stents5. In BVS, LLL also depends on the late scaffold expansion11. Current BVS data show an LLL of 0.16±0.18 mm at 6 months and 0.27±0.20 mm at 2-year follow-up for the second generation of BVS6, while an LLL of 0.21±0.34 mm at 6 months was reported for the DESolve® scaffold (Elixir Medical Corporation, Milpitas, CA, USA)12. Recent analyses have shown that the LLL for the FORTITUDE scaffold is 0.29±0.43 mm at nine months of follow-up, which is comparable with the current BVS previously reported. In our analysis, in-scaffold LLL for the APTITUDE BRS is comparable with the Absorb and FORTITUDE at 9 months (0.35±0.33 mm) and at 24 months (0.37±0.44 mm).
OCT ANALYSIS
The OCT analysis conducted at 9 months showed no statistically significant difference in mean scaffold area (7.82±1.81 mm2 at baseline to 7.84±1.79 mm2 at 9 months). Almost all struts were covered by neointimal tissue (97%) and completely apposed to the vessel wall (96.5±5.02%). A total of 3% of all struts were uncovered but fully apposed. A very low percentage of all struts analysed were covered but malapposed (0.037±0.16%). No uncovered, malapposed struts were detected. Strut apposition and coverage have been important predictors of late stent thrombosis in DES trials. In our study, the high percentage of strut apposition (~99%) and very low percentage of uncovered malapposed struts may result in an improvement in long-term clinical outcomes. However, these OCT findings indicate stent struts still present at nine months, indicating the active resorption process still ongoing. These OCT findings observed during the active process of resorption need to be confirmed in the long term with the use of serial imaging.
CLINICAL OUTCOMES
Serruys et al6 reported a MACE rate at one year of 7.1% for the second generation of BRS in the ABSORB cohort B trial. In the FIM DESolve scaffold study, the overall MACE rate was 20% at one-year follow-up12. RENASCENT II showed very high clinical device and procedural success rates with no MACE reported at hospital discharge. Two non-Q-wave MIs (TV-MIs) were reported because of troponin rise but without ECG changes or clinical symptoms at 9-month follow-up without any TLR. No cardiac death or stent thrombosis was seen at 9-month follow-up. Our analysis demonstrated that the APTITUDE BRS is safe and effective for use in the treatment of de novo stenotic native coronary artery lesions in patients undergoing elective PCI.
Furthermore, there are other new BRS at various stages of testing. These all need to undergo FIM trials and then eventual RCTs with current DES to evaluate their safety and clinical performance13.
Limitations
This study is limited by the number of patients and also the follow-up period. It would also be worth noting that BRS implantation during RENASCENT II was guided by IVUS and OCT assistance. Further studies are required to analyse the results of the APTITUDE BRS using routine implantation techniques as well as assessing clinical outcomes in longer follow-up.
Conclusions
The 24-month clinical experience with the PLLA APTITUDE BRS has demonstrated that the polymer is safe and effective in improving coronary luminal diameter in patients undergoing elective PCI. The APTITUDE BRS has shown that, despite reduction in struct thickness, it matches previous safety clinical endpoints seen with the FORTITUDE BRS.
|
Impact on daily practice RENASCENT II was a first-in-human study to analyse the APTITUDE BRS which was found to be safe and effective up to 24 months. It had low levels of target vessel failure and late lumen loss, warranting further BRS studies with longer follow-up and implantation using standard implantation techniques. |
Appendix. Study collaborators
Matteo Montorfano, MD; IRCCS, San -Raffaele Hospital, Milan, Italy. Hector Hernandez, MD; Interventional Cardiology Unit, Universidad Industrial de Santander, -Bucaramanga, Santander, Colombia. Camilo Arana, MD; Angiografia De Occidente S.A., Cali, Colombia. Antonio Dager, MD; Angiografia De Occidente S.A., Cali, Colombia. Francesco Bedogni, MD; IRCCS Policlinico San Donato, Milan, Italy. Eugenio Stabile, MD; Division of Cardiology, Department of Advanced Biomedical Sciences, University of Naples “Federico II”, Naples, Italy. Mauro De Benedictis, MD; Interventional Cardiology Unit, A.O. Ordine Mauriziano Umberto I, Turin, Italy. Emanuele Meliga, MD; Interventional Cardiology Unit, A.O. Ordine Mauriziano Umberto I, Turin, Italy. Giuseppe Tarantini, MD; Department of Cardiac, Thoracic and Vascular Sciences, University of Padua Medical School, Padua, Italy. David Antoniucci, MD; A.O. Carreggi, Florence, and Division of Cardiology, Ferrarotto Hospital, University of -Catania, Catania, Italy. Alessio La Manna, MD; Division of Cardiology, Ferrarotto Hospital, University of Catania, Catania, Italy. Corrado Tamburino, MD; Division of Cardiology, Ferrarotto Hospital, University of Catania, Catania, Italy.
Funding
This study was funded by Amaranth Medical Inc.
Conflict of interest statement
A. Chieffo reports proctorship fees from Amaranth Medical Inc. J.F. Granada reports being a scientific advisor for and equity shareholder in Amaranth Medical Inc. The other authors have no conflicts of interest to declare.
Supplementary data
To read the full content of this article, please download the PDF.
