
Abstract
Aims: The study sought to investigate clinical and multimodality imaging assessment of a bioresorbable sirolimus-eluting scaffold (NeoVas, Lepu Medical, Beijing, China) for patients with single de novo coronary artery lesions.
Methods and results: The NeoVas first-in-man study was a prospective, open-label study which enrolled 31 patients with single de novo lesions treated with a bioresorbable sirolimus-eluting scaffold. The primary endpoint was target lesion failure (TLF), a composite of cardiac death, target vessel myocardial infarction and clinically indicated target lesion revascularisation (TLR). Angiography, intravascular ultrasound (IVUS) and optical coherence tomography (OCT) imaging were performed at baseline and six months. Procedural success and device success were 100% (31/31 patients). At six months, the rate of TLF was 3.2%, with only one patient having clinically indicated TLR. No scaffold thrombosis was observed. The angiographic six-month in-scaffold late loss was 0.26±0.32 mm. The minimal scaffold area decreased from 7.11±1.56 mm2 post procedure to 6.74±1.38 mm2 at six months, as measured by IVUS. The OCT results showed that the neointimal hyperplasia area was low (1.56±0.46 mm2), with a high proportion of scaffold strut coverage (95.7%).
Conclusions: This first-in-man study shows feasibility, promising clinical and multimodality imaging results up to six months for the NeoVas bioresorbable sirolimus-eluting scaffold in the treatment of patients with simple de novo lesions, with an acceptable in-scaffold late loss, low neointimal hyperplasia, and a high percentage of scaffold strut coverage. (ClinicalTrials.gov Identifier: NCT02195414)
Abbreviations
BRS: bioresorbable scaffold
DES: drug-eluting stent
IVUS: intravascular ultrasound
LLL: late lumen loss
NIH: neointimal hyperplasia
OCT: optical coherence tomography
PLLA: poly-L-lactic acid
QCA: quantitative coronary angiography
TLF: target lesion failure
TLR: target lesion revascularisation
Introduction
Percutaneous coronary intervention (PCI) is a predominant therapeutic modality for patients with coronary artery disease. Clinical trials and real-world registries have confirmed that the use of newer-generation drug-eluting stents (DES), compared with first-generation DES, significantly improves long-term clinical safety and efficacy1. However, metallic stents have inherent limitations, namely the potential risk of neoatherosclerosis, preclusion of future surgical revascularisation, and impairment of the physiological vasomotor function of the stented segment2,3.
In the past decade, tremendous efforts have been made to develop fully bioresorbable devices to provide temporary vessel scaffolding which then gradually disappears. The concept of vascular restoration therapy with a fully bioresorbable scaffold (BRS) has been highlighted recently. In the ABSORB Cohort A study, Ormiston et al demonstrated the feasibility of the first Conformité Européenne mark-approved BRS (Absorb BVS; Abbott Vascular, Santa Clara, CA, USA) in the treatment of patients with simple de novo coronary artery disease4. To date, several poly-L-lactic acid (PLLA)-based polymeric BRSs have been clinically evaluated5. The initial clinical performance of these devices is promising in low- and relatively moderate-risk patients with coronary artery disease6,7.
The NeoVas™ sirolimus-eluting bioresorbable coronary scaffold system (Lepu Medical Technology [Beijing Co., Ltd.], Beijing, China) is a balloon-expandable device consisting of a polymer backbone of PLLA coated with a layer of a 1:1 mixture of poly(D,L-lactide) and the antiproliferative drug sirolimus. The present study was designed to investigate the feasibility, initial safety and efficacy of this novel BRS in the treatment of patients with single de novo coronary lesions.
Methods
STUDY DESIGN AND POPULATION
The NeoVas first-in-man trial is a prospective, two-centre, open-label study that investigated the feasibility, initial safety and efficacy of the NeoVas bioresorbable scaffold in the treatment of patients with simple coronary artery disease. The protocol was approved by the institutional review board and ethics committees at The General Hospital of Shenyang Military Region (Shenyang, China) and the Sir Run Run Shaw Hospital (Hangzhou, China). All patients provided written informed consent for participation in the study. The primary investigators have full access to and take full responsibility for the integrity of the data. All authors have read, and agreed to the manuscript as written.
From July to September 2014, patients with single de novo native coronary artery lesions were eligible for enrolment in the study if they were over 18 years with a diagnosis of stable, unstable, or silent ischaemia and intended to undergo PCI. Target lesions were required to have a visually estimated diameter stenosis of ≥70% and <100%, reference vessel diameter between 2.75 mm and 3.75 mm by online quantitative coronary angiography (QCA) analysis, and a visual lesion length ≤20 mm. The major exclusion criteria included patients with acute myocardial infarction within one month, left ventricular ejection fraction ≤40%, estimated glomerular filtration rate <60 ml/min, chronic total occlusion lesions, in-stent restenosis lesions, thrombotic lesions, ostial lesions, bifurcation lesions involving a side branch ≥2.0 mm, and lesions located in the left main coronary artery.
The main analysis was performed on a per protocol intention-to-treat patient population. One patient with ostial stenosis of the left anterior descending artery (a protocol deviation) was enrolled and included in the analysis. Clinical endpoints were assessed at 30 days and six months (with planned future follow-up annually up to five years). Angiography, intravascular ultrasound (IVUS), and optical coherence tomography (OCT) examinations were conducted at baseline and six-month follow-up.
The NeoVas first-in-man trial is registered at ClinicalTrials.gov NCT02195414.
STUDY DEVICE
The NeoVas scaffold (Lepu Medical) is a balloon-expandable BRS, which consists of four components: a PLLA platform, poly(D,L-lactide) polymer, the antiproliferative drug sirolimus (15.3 μg/mm scaffold lengths), and radiopaque markers at the ends (Figure 1). The total strut thickness of the NeoVas scaffold is 170 μm which consists of a backbone thickness of 160 μm and a polymer thickness of 10 μm.

Figure 1. The NeoVas bioresorbable scaffold, a balloon-expandable, poly-L-lactic acid-based sirolimus-eluting scaffold.
The crossing profile of the device is 1.40 mm and it is 6 Fr compatible. The kinetic release data indicated that about 75% of the drug releases within one month after scaffold implantation. The scaffold sizes used in the present study were 3.0 and 3.5 mm in diameter and 12, 15, 18, and 24 mm in length.
The NeoVas scaffold has to be kept at a temperature between 0 and 10° to ensure device stability and a shelf life of up to one year.
DEFINITIONS
Clinical device success was defined as successful delivery and deployment of the studied scaffold at the targeted lesions with attainment of <50% residual stenosis by visual assessment. Bail-out stenting was not considered as a device failure. Clinical procedure success was defined as attainment of <50% residual stenosis, Thrombolysis In Myocardial Infarction (TIMI) 3 flow and no residual dissection or thrombosis of the target lesion, without the occurrence of major adverse cardiac events up to seven days after the index procedure.
Target lesion failure (TLF) was defined as cardiac death, target vessel myocardial infarction, and/or clinically indicated TLR. Myocardial infarction (Q-wave, non-Q-wave) per protocol was defined as the development of new pathological Q-waves or creatine kinase rise of ≥2 times the upper limit of normal accompanied by creatine kinase-MB rise. Scaffold thrombosis was defined according to the Academic Research Consortium classification8.
STUDY PROCEDURE
Lesions were treated by standard interventional techniques, and predilation with a non-compliant balloon in diameters of 0.5 mm less than the reference vessel diameter was mandatory. The NeoVas scaffold should be deployed stepwise in 2 atm increments every five seconds until at least 9 atm and up to the maximum desired pressure (no more than the rated burst pressure), maintaining the scaffold balloon pressure for at least 30 seconds. Post-dilation after the scaffold implantation was recommended but left to the discretion of the physicians. In case of a bail-out stent and an additional stent being required, the stent had to be the XIENCE V® everolimus-eluting stent (Abbott Vascular, Santa Clara, CA, USA). All patients were pre-treated with aspirin and clopidogrel. Patients who were not pre-treated received 300 mg aspirin and a 300-600 mg loading dose of clopidogrel at least six hours before the index procedure. Heparin was administered before the procedure to maintain an activated clotting time between 250 and 350 seconds. After the procedure, all patients received 100-300 mg per day of aspirin for one month, followed by 100 mg per day indefinitely, as well as clopidogrel 75 mg per day for at least 12 months.
QCA analysis was performed by an independent angiographic core laboratory (China Cardiovascular Research Foundation, Beijing, China) using the software QAngio® XA, version 7.3 (Medis medical imaging systems, Leiden, The Netherlands). The scaffold and peri-scaffold segments of 5 mm proximal and distal to the scaffold edge were analysed. The following parameters for QCA were measured or calculated: reference vessel diameter, minimal lumen diameter, diameter stenosis, acute gain, late lumen loss (LLL), and binary restenosis. Acute recoil was evaluated in angiography as the difference between the mean luminal diameter of the scaffolded vessel and the diameter at maximal balloon inflation. Results are presented as paired matched views post procedure and at six-month follow-up.
IVUS examinations were undertaken with a mechanical catheter (Atlantis™ SR Pro; Boston Scientific, Marlborough, MA, USA) at an automated pullback speed of 0.5 mm/s9. The region of interest included the 5 mm distal segment, in-scaffold segment and 5 mm proximal segment. Vessel, lumen and scaffold areas were measured according to the methods described by Garcia-Garcia et al10. All cross-sectional IVUS images of the stented segments were analysed at 1 mm intervals by an independent core laboratory (FuWai Hospital, Beijing, China), using validated software (QIvus version 2.2; Medis medical imaging systems). The following parameters were analysed: mean vessel area, mean lumen area, minimal lumen area, mean scaffold area, minimal scaffold area, mean neointimal hyperplasia (NIH) area, and in-scaffold area obstruction.
OCT acquisitions were performed using the C7XR Fourier-domain system (St. Jude Medical, St. Paul, MN, USA). After confirming the proper position of the OCT catheter, at least 100 μg nitroglycerine was administered. OCT images were acquired at frame rates of 100 frames/s with pullback speeds of 20 mm/s. Cross-sectional OCT images were analysed at 0.4 mm intervals by an independent core laboratory (FuWai Hospital, Beijing, China), using the software QIvus version 2.2 (Medis). OCT parameters were calculated and defined as follows10. Neointimal thickness was measured as the perpendicular distance between the endoluminal surfaces of the neointima and the scaffold strut. The absence of definite neointima over the scaffold strut was classified as an uncovered scaffold strut. Scaffold strut malapposition was defined as the absence of contact between the strut and vessel wall. Scaffold area was delineated on the abluminal side of the struts and was identical to the lumen area in case of no scaffold malapposition and tissue prolapse. NIH area was defined as the difference between scaffold area and luminal cross-sectional area. Cross-sections located over the side branches more than 1.5 mm were excluded from the analysis.
STATISTICAL ANALYSIS
This first-in-man trial was designed to test the feasibility of the novel NeoVas BRS in the treatment of patients with simple de novo lesions and to generate hypotheses for a future randomised controlled trial. There was no formal sample size calculation for an angiographic or clinical endpoint. The endpoint analyses presented in this report were performed on the basis of both an intention-to-treat population and a per protocol set. Continuous variables are expressed as mean and standard deviation or median and interquartile range depending on the distribution of the data. Categorical variables are shown as counts and percentages. Paired comparisons between post procedure and six-month follow-up were conducted with a Wilcoxon’s signed-rank test. All analyses were undertaken using SPSS, Version 20.0 (IBM Corp., Armonk, NY, USA).
Results
A total of 31 patients with single de novo lesions were enrolled in the NeoVas first-in-man trial. All patients received the studied device and no bail-out stent was needed. The study profile is shown in Figure 2 and baseline characteristics are presented in Table 1. The clinical presentation before scaffold implantation was of stable angina in 29.0% and unstable angina in 71.0%. No complications occurred during the procedure. Clinical device success and procedure success were both 100%.
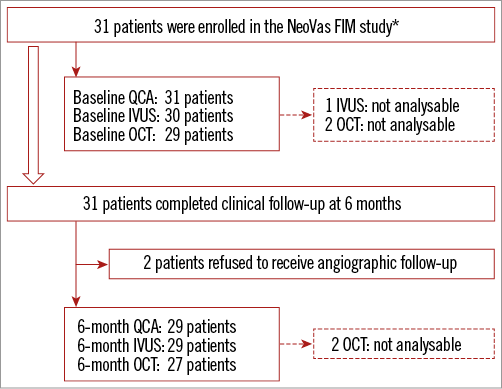
Figure 2. Flow chart of the NeoVas first-in-man trial. *indicates that 1 patient with ostial stenosis of the left anterior descending artery (a protocol deviation) was enrolled. FIM: first-in-man; IVUS: intravascular ultrasound; OCT: optical coherence tomography; QCA: quantitative coronary angiography
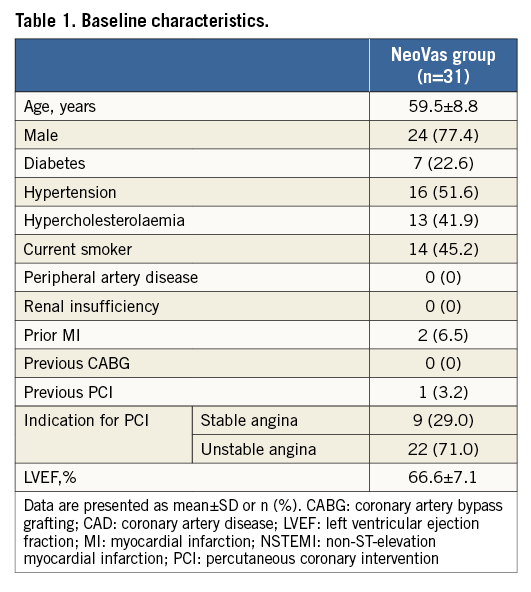
Lesion and procedural characteristics are presented in Table 2; 96.6% of patients had B1/B2 lesions according to the ACC/AHA lesion classification. Angiographic acute recoil was 0.15±0.07 mm. The majority of patients (96.8%) had balloon dilation after device implantation, with a nominal diameter of 3.45±0.30 mm and maximum pressure of 18.4±3.66 atm. Angiographic follow-up at six months was completed in 93.5% of patients. In the intention-to-treat population, the acute gain was 1.76±0.48 mm, and the mean in-scaffold LLL was 0.26±0.32 mm (Table 3, Figure 3). In the per protocol set, the acute gain was 1.77±0.49 mm, and the mean in-scaffold LLL was 0.20±0.12 mm.
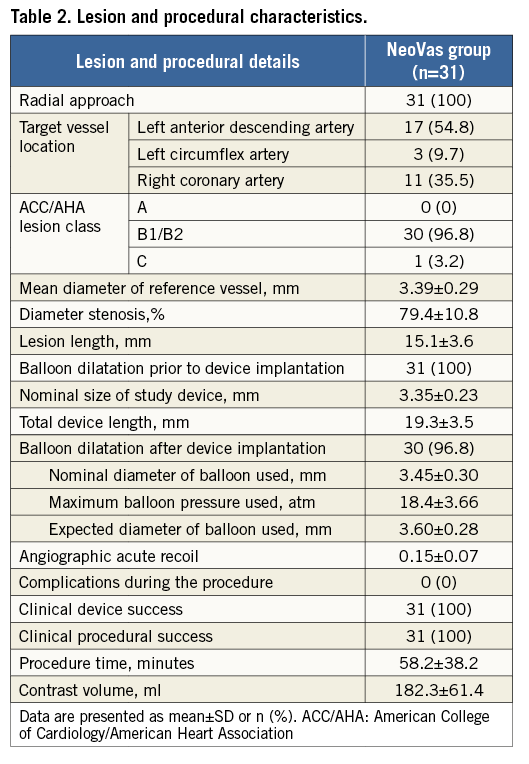
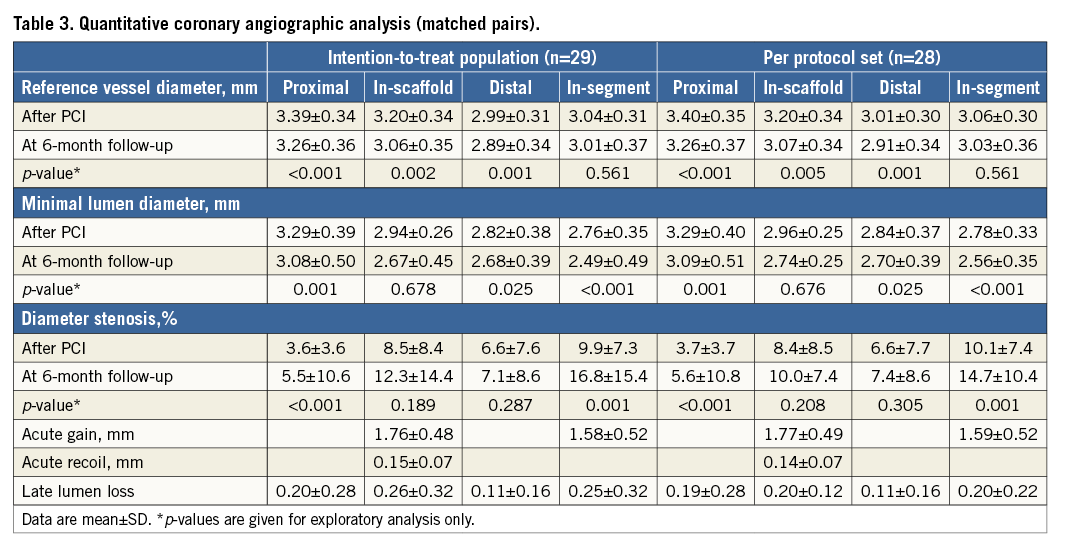
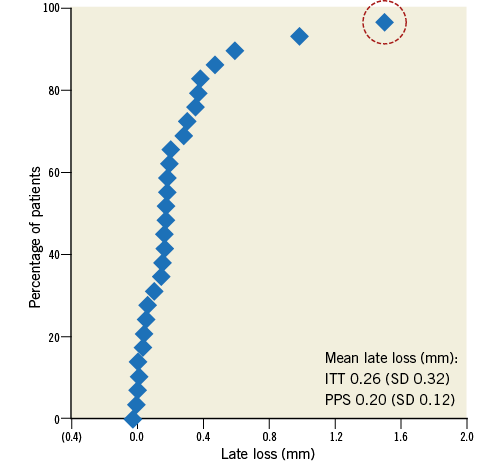
Figure 3. Cumulative frequency distribution curve of in-scaffold late loss. The dashed circle indicates the case (a protocol deviation) which received reintervention at 187 days (late loss: 1.5 mm). ITT: intention-to-treat; PPS: per protocol set
Serial IVUS examinations demonstrated a numerical decrease in the mean scaffold cross-sectional area (8.49±1.67 mm2 post procedure vs. 8.36±1.53 mm2 at six months, p=0.474) (Table 4), showing the absence of late scaffold recoil. A significant reduction was noted in the minimal lumen area, whilst the mean lumen area remained unchanged between post procedure and six-month follow-up, suggesting a good neointimal inhibition of the drug (in-scaffold area obstruction: 3.98% [3.25%-5.34%]).
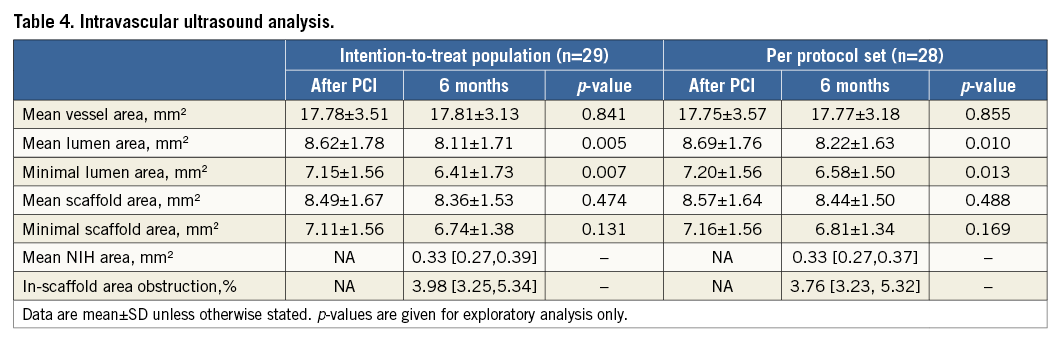
Table 5 presents the analysis of OCT on both the strut level and cross-sectional level. A representative case with OCT post procedure and at six-month follow-up is shown in Figure 4. The scaffold struts were well covered by a thin, uniform layer of neointima (mean thickness of strut coverage: 0.08±0.06 mm) in 95.7% of patients. For the analysis of malapposed struts, a significant decrease was noted for the proportion of malapposed struts from baseline (4.0%) to six-month follow-up (0.58%). Consistent with the IVUS results, the mean NIH area was low (1.56±0.46 mm2).
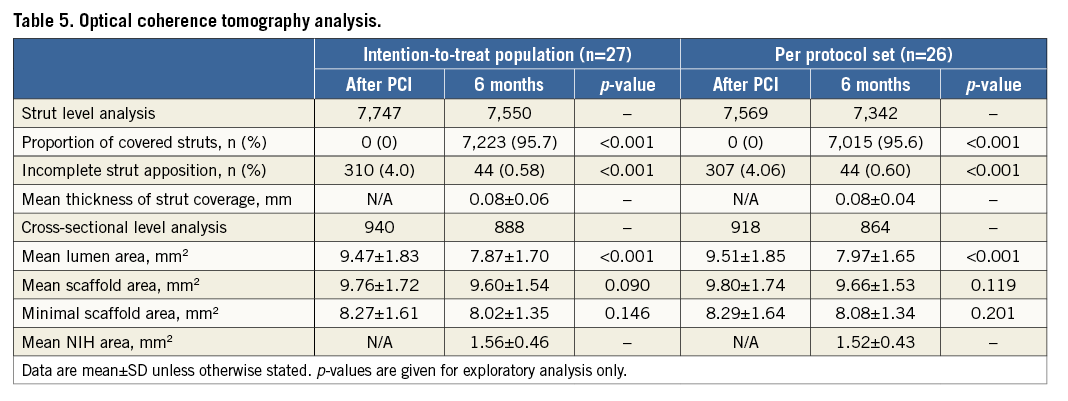
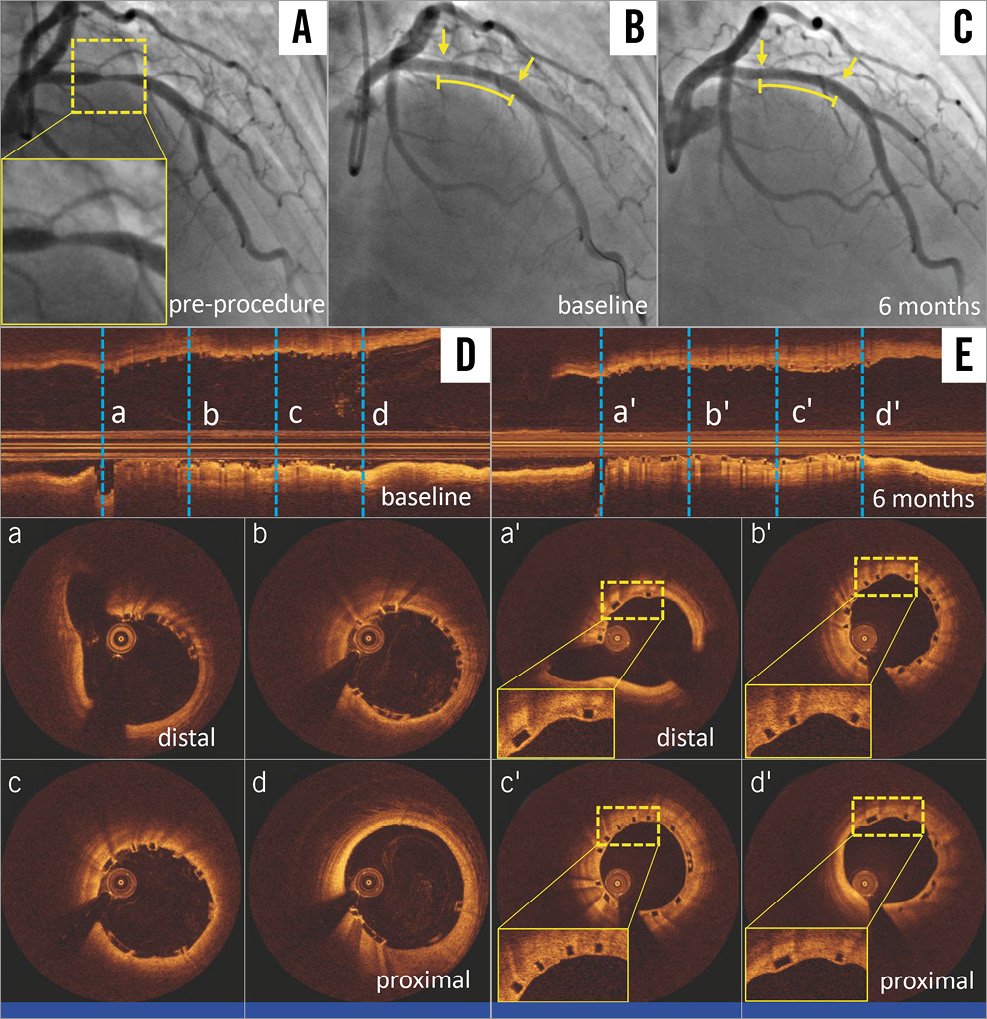
Figure 4. A representative case treated with the NeoVas bioresorbable scaffold. Target lesion located in left anterior descending artery (A). Repeat angiography post NeoVas scaffold implantation (B). Angiography at six-month follow-up (C). OCT longitudinal views post procedure (D) and at six-month follow-up (E). OCT cross-sectional views post procedure (a-d) and at six-month follow-up (a’-d’). The curved lines and arrows indicate the scaffolded segments. OCT: optical coherence tomography
At six months, clinical follow-up was completed in 100% of patients. Only one patient experienced clinically indicated TLR, which resulted in a 3.23% incidence of TLF. There was no cardiac death, myocardial infarction or scaffold thrombosis within six months after the index procedure.
Discussion
The study was a prospective, single-arm, open-label clinical trial evaluating the feasibility, initial safety and efficacy of the NeoVas PLLA-based sirolimus-eluting bioresorbable coronary scaffold system in the treatment of patients with single de novo lesions. This first-in-man trial has shown that the NeoVas scaffold can be successfully implanted without any significant acute recoil, and effectively inhibited NIH with a low angiographic in-scaffold LLL (0.26±0.32 mm) at six months. IVUS demonstrated uniform coverage of neointima and low in-scaffold area obstruction (3.98%). The device showed early vascular healing with achievement of a high proportion of scaffold strut coverage (95.7%) and low incomplete strut apposition (0.58%). Only one patient had a major adverse cardiac event (clinically indicated TLR), and no scaffold thrombosis occurred up to six months.
Several BRSs with different designs are now available in China2. Some of these devices have been clinically tested. The ABSORB China trial, comparing the Absorb BVS against the metallic everolimus-eluting stent XIENCE V, demonstrated the non-inferiority of the Absorb BVS to XIENCE V in terms of one-year in-segment LLL (0.19±0.38 mm vs. 0.13±0.38 mm, p for non-inferiority=0.01)11. One-year clinical results showed that Absorb BVS and XIENCE V also had similar incidences of target lesion failure (3.4% vs. 4.2%, p=0.62) and definite/probable scaffold thrombosis (0.4% vs. 0.0%, p=1.00). The Xinsorb BRS (Huaan Biotechnology, Shanghai, China) is a PLLA-based polymer scaffold and contains the antiproliferative drug sirolimus12. An initial preclinical study showed that this novel BRS was safe and feasible in the prevention of NIH within one month. Currently, the device is still under clinical evaluation. In the present study, we demonstrated for the first time the feasibility of the novel NeoVas scaffold for patients with simple coronary artery disease. Both clinical device and procedural success were 100%. A further randomised clinical trial (ClinicalTrials.gov Identifier: NCT02305485) comparing the clinical safety and efficacy of the NeoVas scaffold with the XIENCE™ (Abbott Vascular, Santa Clara, CA, USA) everolimus-eluting stent was launched in 2015. The detailed progress of the trial is expected to be updated in the near future.
The main function of metallic stents is to scaffold the vessel wall and prevent early elastic recoil and acute vessel closure. In previous human clinical trials, acute stent recoil varied between 3% and 15% following metallic stent implantation. This wide range was partially associated with stent material and design and the difference in definitions of recoil13,14. Our study demonstrated that the acute scaffold recoil, calculated as the difference between the mean diameter of the last inflated balloon and the mean luminal diameter immediately after the last balloon deflation, was low (0.15±0.07 mm), similar to that of the Absorb BVS.
Although PLLA-based BRSs have thicker scaffold struts to maintain sufficient radial strength compared with DES made from metallic alloys, the relationship between the thick scaffold struts of BRSs and periprocedural complications, such as side branch occlusion, seems weak. In the ABSORB II trial, Ishibashi et al showed that there were no differences in the incidence of creatine kinase rise and periprocedural myocardial infarction between the Absorb BVS and an everolimus-eluting stent15. In the present study, we did not find periprocedural myocardial infarction post NeoVas scaffold implantation, even though the studied device (170 μm) has a relatively thicker strut than other BRSs (Absorb BVS: 157 μm, DESolve scaffold: 150 μm).
In our study, late luminal loss of the NeoVas scaffold was 0.26 mm at six-month follow-up. This value is better than the Absorb BVS 1.0 (LLL 0.44 mm)4, but higher than that of Absorb BVS version 1.1 (LLL 0.16 mm) at six-month follow-up16. This can be explained partially by the fact that the effect and loads of antiproliferative drugs in these scaffolds are different. However, the mean scaffold areas of the NeoVas scaffold by IVUS and OCT remained unchanged between baseline and follow-up, suggestive of the absence of late recoil. The extent of late recoil is the result of balance between the elastic recoil and the radial strength of the scaffold, along with the elastic properties of the arterial wall and the plaque characteristics of the scaffolded segments. Therefore, further studies in patients with complex lesions will be needed to confirm lack of late recoil.
The vascular healing response of a coronary stent platform post implantation may guide the duration of dual antiplatelet therapy in individual patients. Clinical studies have raised concerns about late and very late stent thrombosis if vascular healing was delayed with the presence of a number of uncovered or malapposed stent struts post device implantation17-19. In the ABSORB Cohort B1 study, the coverage of struts at six months was almost complete, with only 2.04% of struts remaining uncovered16. Incomplete strut apposition was seen in 11 scaffolds at six months, but only one scaffold with strut malapposition was detected at two years. Similarly, another CE-approved BRS, the DESolve® (Elixir Medical Corporation, Sunnyvale, CA, USA), also had a very high frequency of covered struts per scaffold (98.7%) at the same follow-up period7. Our OCT results showed that early vascular healing was achieved in patients undergoing NeoVas implantation, with a high proportion of covered struts (95.7%) and a low incidence of incomplete strut apposition (0.58%) at six months. Compared with other scaffolds, the strut coverage of the NeoVas scaffold is numerically low, which can be attributed to the relatively thicker scaffold struts of the NeoVas scaffold. However, it is important to note that vascular healing is a multifactorial process and may partly be due to the differences of not just these devices, but also of lesions and patients.
Meta-analyses have suggested increased definite/probable scaffold thrombosis in patients undergoing Absorb BVS implantation compared with an everolimus-eluting stent, but no significant difference in mortality and TLF20,21. Furthermore, patients treated with Absorb BVS had the highest risk of definite/probable scaffold thrombosis between one and 30 days after implantation (odds ratio: 3.11, 95% CI: 1.24-7.82, p=0.02). This could potentially be attributed to implantation technique and lesion selection. In an all-comers registry, Wiebe et al reported that the first 100 consecutive patients treated with Absorb BVS had significantly high incidences of target vessel failure (1.1% vs. 10.1%, p<0.01) compared with the next 100 patients during a follow-up of 210 days22. Furthermore, the implantation of BRS is different from that of metallic stents due to different mechanical properties. A survey from 14 European centres with high volumes of BVS procedures proposed that current BVS devices require special procedural considerations to achieve optimal short- and long-term results23. Compared to experiences with the Absorb BVS, post-dilation was used significantly more often in the present study (96.8%). However, it still remains uncertain how the procedural behaviour influences early clinical outcomes after BRS implantation. In our study, the incidence of major adverse cardiac events was low, with only one patient undergoing revascularisation related to the target lesion. Even though no scaffold thrombosis occurred in patients treated with the NeoVas scaffold during this observational period, it is impossible to reach any conclusion with respect to the clinical safety of the NeoVas scaffold for treating patients with coronary artery disease. Therefore, appropriately powered randomised trials are necessary to confirm our results and evaluate the clinical performance of the NeoVas scaffold.
Limitations
The NeoVas first-in-man study was an exploratory observational study to investigate a novel technology; thus, no special sample size calculation was performed. The follow-up duration is relatively short and not adequate to assess scaffold bioresorption and potential strengths. Intravascular imaging modalities have suggested good safety and efficacy of the NeoVas scaffold for treating patients with simple lesions. However, the results of the trial should be interpreted with caution and considered hypothesis-generating and of unclear clinical relevance.
Conclusions
This first-in-man trial was designed to evaluate the feasibility and initial safety and efficacy of the novel NeoVas scaffold in the treatment of patients with single de novo lesions. A few clinical events were recorded within six months post NeoVas scaffold implantation. IVUS and OCT examinations showed good early vascular healing and acceptable LLL with the NeoVas scaffold. Large-scale randomised trials and registries are warranted to assess the clinical performance of the NeoVas scaffold in patients with moderately complex lesions or scenarios.
| Impact on daily practice Several PLLA-based polymeric bioresorbable scaffolds have been clinically evaluated. The NeoVas scaffold is a balloon-expandable BRS consisting of four components: a PLLA platform, poly(D,L-lactide) polymer, sirolimus, and radiopaque markers at the ends. This first-in-man study has shown the feasibility, initial safety and efficacy of the NeoVas PLLA-based sirolimus-eluting bioresorbable coronary scaffold system in the treatment of patients with single de novo lesions. Certainly, appropriately powered randomised trials are necessary to evaluate the clinical performance of the NeoVas scaffold in patients with moderately complex lesions or scenarios. |
Acknowledgements
The authors express their gratitude to all participants in the NeoVas first-in-man study, whose work made this study possible.
Funding
The NeoVas FIM trial was sponsored by Lepu Medical.
Conflict of interest statement
The authors have no conflicts of interest to declare.
