Abstract
Dual antiplatelet therapy (DAPT) is currently the standard of care after percutaneous coronary intervention (PCI). Recent studies suggest that reducing DAPT to 1-3 months followed by an aspirin-free single antiplatelet therapy (SAPT) strategy with a potent P2Y12 inhibitor is safe and associated with less bleeding. However, to date, no randomised trial has tested the impact of initiating SAPT immediately after PCI, particularly in patients with acute coronary syndromes (ACS). NEOMINDSET is a multicentre, randomised, open-label trial with a blinded outcome assessment designed to compare SAPT versus DAPT in 3,400 ACS patients undergoing PCI with the latest-generation drug-eluting stents (DES). After successful PCI and up to 4 days following hospital admission, patients are randomised to receive SAPT with a potent P2Y12 inhibitor (ticagrelor or prasugrel) or DAPT (aspirin plus a potent P2Y12 inhibitor) for 12 months. Aspirin is discontinued immediately after randomisation in the SAPT group. The choice between ticagrelor and prasugrel is at the investigator’s discretion. The primary hypothesis is that SAPT will be non-inferior to DAPT with respect to the composite endpoint of all-cause mortality, stroke, myocardial infarction or urgent target vessel revascularisation, but superior to DAPT on rates of bleeding defined by Bleeding Academic Research Consortium 2, 3 or 5 criteria. NEOMINDSET is the first study that is specifically designed to test SAPT versus DAPT immediately following PCI with DES in ACS patients. This trial will provide important insights on the efficacy and safety of withdrawing aspirin in the early phase of ACS. (ClinicalTrials.gov: NCT04360720)
Introduction
Acute coronary syndrome (ACS) guidelines suggest that antithrombotic therapies should be tailored to an individual’s haemorrhagic and ischaemic risk123. Single antiplatelet therapy (SAPT) strategies have been proposed that reduce bleeding complications following percutaneous coronary intervention (PCI) without increasing the risk of cardiovascular events4567891011. Most trials have shown that either acetylsalicylic acid (ASA) or P2Y12 receptor antagonists can be safely withdrawn after at least one month of dual antiplatelet therapy (DAPT). The potential benefit of dropping ASA in the early phase of ACS, when more potent P2Y12 inhibitors (prasugrel or ticagrelor) are in place, is attractive, especially in terms of preventing gastrointestinal bleeding. However, the optimal timing for ASA withdrawal is still to be determined.
SAPT with a potent P2Y12 receptor antagonist, without ASA, may reduce the risk of bleeding while preserving efficacy regarding ischaemic events when compared with traditional DAPT shortly after PCI in ACS patients. To test this hypothesis, we designed the “PercutaNEOus coronary intervention followed by Monotherapy INstead of Dual antiplatelet therapy in the SETting of acute coronary syndromes: The NEO-MINDSET Trial" (NEOMINDSET) to evaluate the efficacy and safety of withdrawing ASA immediately after PCI with the latest generation of drug-eluting stents (DES) in ACS patients (ClinicalTrials.gov: NCT04360720).
Methods
Study organisation and design
NEOMINDSET is an investigator-initiated, multicentre, randomised, open-label trial with blinded outcome assessment funded by the Institutional Development Support Program of the Brazilian Unified Health System (PROADI-SUS). The trial is ongoing and aims to test the comparative efficacy and safety of SAPT with a potent P2Y12 inhibitor versus DAPT with ASA plus a potent P2Y12 inhibitor in ACS patients undergoing PCI with DES (Figure 1). The executive committee is responsible for the scientific and operational conduct of the study, integrity of data analysis and reporting of results. The trial is being conducted in accordance with the Good Clinical Practice guidelines. Ethical committees at each participating site have approved the study protocol, and all patients must provide informed consent before enrolment in the trial.
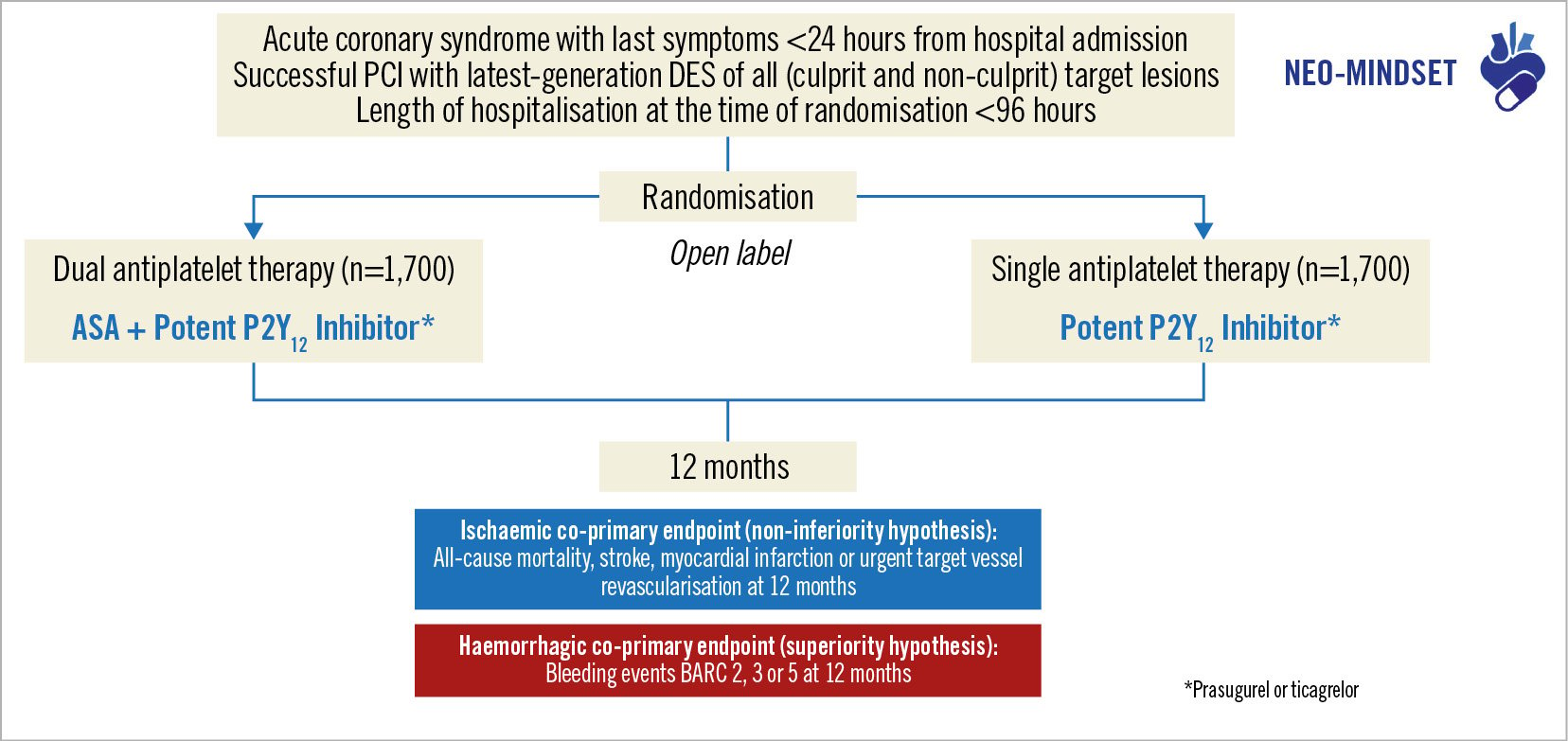
Figure 1. NEOMINDSET Trial design. Patient selection starts after successful percutaneous coronary intervention with latest-generation drug-eluting stents, and patients can be consented within 96 hours of hospital admission due to acute coronary syndromes. Patients will be randomised in a 1:1 ratio to receive dual antiplatelet therapy with aspirin and a potent P2Y12 inhibitor or single antiplatelet therapy with a potent P2Y12 inhibitor. The choice between prasugrel or ticagrelor will be made at the investigator’s discretion. The 2 co-primary endpoints will be assessed at 1-year follow-up. ASA: acetylsalicylic acid; BARC: Bleeding Academic Research Consortium; DES: drug-eluting stent; PCI: percutaneous coronary intervention
Study objective
The primary objective of the NEOMINDSET Trial is to evaluate the efficacy and safety of SAPT with a potent P2Y12 inhibitor compared with traditional DAPT for 12 months following PCI with DES in ACS patients. The primary hypothesis of the study is that SAPT is non-inferior for ischaemic events and superior for bleeding events compared with DAPT.
Study population – enrolment and randomisation
A full list of inclusion and exclusion criteria are presented in Table 1. Briefly, we will include 3,400 patients aged 18 years or older with atherosclerotic coronary disease presenting with ST-elevation or non-ST-elevation ACS, whose last symptoms occurred within 24 hours prior to hospital admission. To be included, patients must undergo successful PCI of one or more coronary artery stenoses with DES implantation (culprit and non-culprit vessels) within 96 hours of hospitalisation and should have no further planned additional revascularisation. Successful PCI is determined per the treating physician based on criteria relating to the combination of optimum stent results in the target lesion(s) (no significant residual intra-stent or border residual stenosis or dissection and no occurrence of thrombosis, distal embolism, occlusion of a major side branch, or “no-reflow” phenomenon). Patients undergoing staged PCI are enrolled after completion of all planned procedures; these must be completed within the 96-hour time frame from hospital admission.
Key exclusion criteria are the following: ACS treated conservatively, surgically or with unsuccessful PCI; an intended DAPT duration of less than 12 months; presence of residual coronary stenosis with a high likelihood of future revascularisation in the next year; indication for oral anticoagulation; index event of a presumed non-atherothrombotic cause; major active or recent bleeding; ischaemic stroke in the prior month; and any previous haemorrhagic stroke.
Randomisation is performed centrally through an electronic data capture system, in blocks of permutable sizes, stratified by study site.
Table 1. Study enrolment criteria.
| Inclusion criteria |
|---|
| 1. Aged ≥18 years old |
| 2. ACS with last symptoms <24 hours from hospital admission |
| 3. Successful PCI of all target lesions with latest-generation drug-eluting stents (culprit and non-culprit vessels) |
| 4. Hospitalisation for less than 96 hours |
| 5. Signed written informed consent |
| Exclusion criteria |
| 1. ACS at index hospitalisation treated conservatively, with unsuccessful percutaneous coronary intervention, or undergoing coronary artery bypass graft surgery |
| 2. Impossible, according to the judgment of the investigator, to treat the patient with dual antiplatelet therapy for 12 months |
| 3. Presence of residual lesions with a high likelihood of future intervention within the next 12 months |
| 4. Received fibrinolytic therapy <24 hours from randomisation |
| 5. Need for oral anticoagulation with warfarin or non-vitamin K antagonist anticoagulants |
| 6. Chronic bleeding diathesis |
| 7. Major active or recent bleeding (in-hospital) |
| 8. Previous intracranial haemorrhage |
| 9. Ischaemic stroke in the prior 30 days |
| 10. Presence of brain arteriovenous malformation |
| 11. Index event of non-atherothrombotic cause (e.g., stent thrombosis, coronary embolism, spontaneous coronary dissection, myocardial ischaemia due to supply/demand imbalance) |
| 12. Scheduled or potential cardiac or non-cardiac surgery within the next 12 months |
| 13. Platelet count <100,000 cells/mm3 or >700,000 cells/mm3 |
| 14. Total white blood count <3,000 cells/mm3 |
| 15. Suspected or documented active liver disease (including laboratory evidence of hepatitis B or C) |
| 16. Heart transplant recipient |
| 17. Known allergies or intolerance to ASA, clopidogrel, ticlopidine, ticagrelor, prasugrel, heparin or -limus family antiproliferative agents |
| 18. Life expectancy lower than 1 year |
| 19. Any significant medical condition that, in the investigator’s opinion, may interfere with optimal patient participation in this study |
| 20. Participation in another trial within the last 12 months, unless the subject is deemed to benefit from participation in this study |
| ACS: acute coronary syndrome; ASA: acetylsalicylic acid; PCI: percutaneous coronary intervention |
Study treatment and follow-up
After hospital admission for ACS, investigators have up to 96 hours to include the participant in the study. Prior to randomisation, patients can receive any combination of antiplatelet agents as per local practice (Figure 2). If a participant undergoes staged PCI, standard DAPT will be administered between the initial PCI and the staged procedure, as per standard of practice. Randomisation is performed after the last PCI, when the coronary revascularisation plan is considered finished and has been successfully accomplished. The treating physician selects either ticagrelor or prasugrel as the P2Y12 inhibitor to be given immediately after randomisation. Patients are then randomised in an open-label 1:1 fashion to receive low-dose ASA (80-100 mg) plus the chosen P2Y12 antagonist (DAPT group) or to withdraw ASA and maintain monotherapy with only the selected P2Y12 inhibitor (SAPT group) for 12 months. Protocol-mandated therapy begins directly after randomisation, and ASA is discontinued immediately in the SAPT group (Supplementary Figure 1). Details on study treatment administration are presented in Supplementary Appendix 1.
Patients have in-person follow-up visits at 1, 6 and 12 months after randomisation to assess for adverse events, study endpoints and treatment adherence. Telephone calls occur at 3, 9 and 13 months after randomisation for the same purpose. Study drug adherence will be assessed using the Non-adherence Academic Research Consortium (NARC) consensus12. Non-compliance will be categorised per type of non-adherence, the decision-making process, reasons and timing of non-adherence. After 12 months of study treatment, the antiplatelet regimen will be chosen at the investigator’s discretion.

Figure 2. Study protocol timelines. To be included, patients should present with last symptoms occurring within 24 hours of hospital admission. Once hospitalised, investigators will have up to 96 hours to include the participant in the study. Randomisation will be performed after coronary revascularisation is considered complete. A loading dose of prasugrel or ticagrelor will be given immediately after randomisation to those patients who previously received clopidogrel. ACS: acute coronary syndrome; DES: drug-eluting stent; PCI: percutaneous coronary intervention
Study endpoints
Primary endpoint
The co-primary endpoints are an ischaemic endpoint (composed of all-cause mortality, stroke, myocardial infarction or urgent target vessel revascularisation) and a bleeding endpoint (major or clinically relevant non-major bleeding defined by Bleeding Academic Research Consortium [BARC] 2, 3 or 5 criteria)13. In brief, BARC Type 2, 3 and 5 bleeds are defined as follows: clinically overt haemorrhage requiring medical attention (Type 2); requiring transfusion, surgical correction or associated with a haemoglobin drop of at least 3 g/dl (Type 3); or fatal events (Type 5). Detailed definitions of the ischaemic endpoints are presented in Supplementary Appendix 2.
The co-primary endpoints are assessed between randomisation and 12 months (equalling 365 days). All clinical endpoints are adjudicated by an external independent clinical event classification committee, whose members are unaware of the treatment-group assignments.
Secondary endpoints
Secondary endpoints include all-cause death, cardiovascular death, non-cardiovascular death, myocardial infarction, stroke, sudden death at 30 days, stent thrombosis, unplanned invasive coronary intervention, BARC 1-5 bleeding, and net efficacy and safety outcomes. Primary and secondary endpoints are shown in Supplementary Table 1.
Power, sample size and statistical considerations
This trial was designed to test hierarchically whether SAPT as compared with DAPT would be non-inferior with regard to ischaemic events and superior with respect to major or non-major clinically relevant bleeding. The GLASSY study14, a dedicated analysis of adjudicated events from the GLOBAL LEADERS Study6, indicated that the composite of all-cause death, non-fatal MI, non-fatal stroke, or urgent target vessel revascularisation was 9.6% in ACS patients after 2 years of DAPT, with approximately two-thirds of the events occurring during the first year. Based on these assumptions, we calculated that the enrolment of 3,400 patients would provide 80% power to establish non-inferiority regarding ischaemic events, assuming an 7.0% cumulative incidence at 1 year in the DAPT group and an absolute non-inferiority margin of 2.5%. If non-inferiority is demonstrated, the analysis for the bleeding endpoint will be performed to assess the superiority of SAPT over DAPT, with an estimated 88% power of demonstrating superiority with an assumed 8.0% occurrence of bleeding events4 in the DAPT group and 5.0% in the SAPT group (37.5% relative risk reduction). The final sample size takes into account an anticipated 4% loss to follow-up.
Analysis for the co-primary endpoints
All patients will be analysed following the intention-to-treat principle and reported according to their randomised arm, irrespective of the treatment they receive. Cumulative incidences of the co-primary and secondary endpoints within 365 days of randomisation will be estimated using the Kaplan-Meier method. A sensitivity analysis will be performed in the per-protocol population, excluding participants with violations of the inclusion/exclusion criteria at the time of randomisation and/or who did not receive the assigned treatment at the time of randomisation. Follow-up will be censored at the last date of known outcome status or at 12 months from randomisation, whichever comes first.
All adjudicated events occurring from randomisation to 365 days post-randomisation will be considered. A hazard ratio (HR) and 95% confidence interval (CI) for the primary endpoint will be generated using the Cox proportional hazards model that will include the treatment group (SAPT vs DAPT) as a covariate. For the ischaemic endpoint, a 1-sided non-inferiority test with a type 1 error of 0.025 will be performed. The null hypothesis will be rejected if the upper limit of the 95% CI for the absolute difference in risk in 12-month event rates is less than 2.5%. In case non-inferiority for the ischaemic endpoint is demonstrated, the SAPT scheme will be tested for its superiority in reducing the bleeding co-primary endpoint in comparison with DAPT, using a 2-sided alpha level of 0.025. Analyses for the secondary endpoints and for subgroups are presented in Supplementary Appendix 3.
Safety monitoring
An independent data and safety monitoring board (DSMB) is reviewing unblinded patient-level data for safety on an ongoing basis during the trial. A preliminary safety analysis occurred on 20 December 2021 after 400 subjects were enrolled and followed for at least 30 days. The first interim analysis was performed on 30 June 2022 after inclusion and 30-day follow-up of one-third (n=1,133) of the trial population, and the second interim analysis occurred on 19 December 2022 after inclusion and 30-day follow-up of half (n=1,700) of the trial population. The DSMB recommended that the trial continue as planned after these 3 analyses. One more interim analysis will occur after three-fifths (n=2,040) of the sample size have been enrolled and followed for at least 30 days. Additional interim analyses can be convened at the discretion of the DSMB chairperson, especially if new scientific data or concerns are raised during the conduct of the study that justify such a decision. The DSMB members do not have a primary affiliation with the study sponsor or the Steering Committee of the trial. For each interim analysis, the DSMB reviews data regarding the primary and secondary endpoints, as well as adverse events in the 2 study arms. Based on the data, the DSMB may recommend modifications to the protocol, suspension, or termination of the trial to the Steering Committee. All final decisions regarding trial modifications rest with the Steering Committee.
Discussion
The primary hypothesis of the NEOMINDSET Trial is that SAPT with a potent P2Y12 inhibitor (ticagrelor or prasugrel without aspirin) started shortly after successful PCI with DES in ACS patients will provide adequate ischaemic protection while mitigating bleeding risk when compared to traditional DAPT (aspirin plus prasugrel or ticagrelor). This trial differs from other trials investigating antithrombotic strategies in ACS since ASA will be withdrawn in the first 96 hours of hospital admission in the SAPT arm, thus challenging the concept that classic DAPT with ASA is necessary for a minimum of 1-3 months post-PCI for the prevention of recurrent thrombotic events in the very early phase of ACS.
DAPT is commonly indicated for 12 months following hospital admission for ACS. However, current guidelines suggest that patient profiles and procedural features should be used to guide antithrombotic treatment choice and duration2. Indeed, the use of latest-generation DES is associated with lower risks of thrombotic and restenotic complications compared with bare metal stents, which, theoretically, would decrease the need for prolonged treatment with antiplatelet agents1516. Additionally, patients at high bleeding risk are often encountered in daily practice and are a subset for whom the benefits of prolonged DAPT are not well known, due to the fact that these patients are commonly excluded from clinical trials. Thus, it is possible that DAPT may be used for a shorter duration, or even substituted by SAPT, in a substantial number of ACS patients treated with potent P2Y12 inhibitors.
Early scientific evidence for the safety of SAPT was published by Colombo et al17. In their study, using Palmaz-Schatz stent implantation guided by intracoronary ultrasound, 321 patients treated with ticlopidine or aspirin showed optimal final intracoronary ultrasound outcomes. Stent thrombosis rates were 0.8% (2 events) among 252 patients treated with ticlopidine alone and 1.4% (1 event) among 69 patients treated with aspirin alone after stenting. With the technological development of stents with thinner mesh, bioabsorbable polymers and a better balance between antistenotic effects and mesh coverage, it can be hypothesised that stent thrombosis rates would be even lower than those seen with Palmaz-Schatz, making SAPT a safe and attractive option. The findings of the ASET Pilot Study, which showed no acute thrombotic complications in 201 patients treated with SAPT − only prasugrel − after successful PCI with everolimus-eluting stent implantation for stable coronary disease, support this concept18. Additionally, the OPTICA Study tested SAPT with ticagrelor or prasugrel following PCI in 75 patients with non-ST-segment elevation MI, and no stent thrombosis occurred in 6 months19.
Several contemporary trials have tested different antithrombotic strategies following PCI in acute and stable settings2021. NEOMINDSET differs from prior studies in the following aspects: a) this trial is fully dedicated to ACS patients undergoing successful PCI with DES, including both ST-segment elevation and non-ST-segment elevation ACS; b) ASA is dropped in the SAPT group within the first 96 hours of hospital admission; c) all participants are treated with potent P2Y12 inhibitors and the choice between prasugrel or ticagrelor is made at the investigator’s discretion in a pragmatic manner; d) there are no restrictions on stent type, as long as latest-generation DES are used. We believe our results will be applicable to a broad population of ACS patients as it reflects real-world clinical practice. In conclusion, NEOMINDSET will provide important insights on the efficacy and safety of an early aspirin-free strategy − immediately following PCI − in ACS patients treated with potent P2Y12 inhibitors and will help to inform international guidelines and guide clinical practice.
Limitations
The COVID-19 pandemic delayed site activation and participant recruitment in the trial. The first patient was enrolled in NEOMINDSET in October 2020, and enrolment is expected to continue until October 2023. At present, 51 sites have been activated with a total enrolment of 2,900 patients.
Conclusions
The NEOMINDSET Trial is a multicentre, randomised, open-label trial with a blinded outcome assessment designed to compare SAPT versus DAPT in 3,400 ACS patients undergoing PCI with latest-generation DES. This study will provide important insights on the efficacy and safety of withdrawing aspirin in the early phase of ACS.
Funding
This study is supported by the Brazilian Program for Institutional Development of the Unified Healthcare System (PROADI-SUS) (grant number NUP: 25000.164095/2020-71). Dr Lemos is supported, in part, by agrant from The National Council for Scientific and Technological Development (CNPq) - Brazil (grant number 306677/2019-9).
Conflict of interest statement
R.H.M.Furtado reports personal fees from AstraZeneca, Apsen, Bayer, and Servier; and research grants from AstraZeneca, Novartis, Amgen, Brainfarma, the Brazilian Ministry of Health, and the University Health Network. J.C.Nicolau has received research grants from Amgen, AstraZeneca, Bayer, CSL Behring, Daiichi Sankyo, Dalcor, Esperion, Janssen, Novartis, Novo Nordisk, Sanofi, and Vifor; and is the recipient of ascholarship from the Brazilian National Council for Scientific and Technological Development (CNPq #303448/2021-0). R.D.Lopes reports research support from Bristol-Myers Squibb, GlaxoSmithKline, Medtronic, and Pfizer; consulting fees from Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Daiichi Sankyo, GlaxoSmithKline, Medtronic, Merck, Pfizer, and Portola. M.Valgimigli reports personal fees from AstraZeneca; grants and personal fees from Terumo; personal fees from Alvimedica/CID, Abbott Vascular, Daiichi Sankyo, Bayer, CoreFLOW, Idorsia Pharmaceuticals Ltd, Universität Basel, Dept. Klinische Forschung, Vifor, Bristol-Myers Squibb, Biotronik, Boston Scientific, Medtronic, Vesalio, Novartis, Chiesi, and PhaseBio, outside the submitted work.D.J. Angiolillo reports consulting fees or honoraria from Abbott, Amgen, AstraZeneca, Bayer, Biosensors, Boehringer Ingelheim, Bristol-Myers Squibb, Chiesi, Daiichi Sankyo, Eli Lilly, Haemonetics, Janssen, Merck, Novartis, PhaseBio, PLx Pharma, Pfizer, Sanofi, and Ventura, outside the present work. D.J. Angiolillo also declares that his institution has received research grants from Amgen, AstraZeneca, Bayer, Biosensors, CeloNova, CSL Behring, Daiichi Sankyo, Eisai, Eli Lilly, Gilead, Janssen, Matsutani Chemical Industry Co., Merck, Novartis, Osprey Medical, Renal Guard Solutions, and the Scott R. MacKenzie Foundation. P.W. Serruys declares consultancy for SMT, Philips, Novartis, Meril Life, and Xeltis. O.Berwanger reports consultancy fees from Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Novartis Pharma, and Pfizer. P.A.Lemos reports institutional research funding from, and/or being an unpaid advisory board member, and/or unpaid member of the steering/executive/data safety and monitoring group of trials, and/or unpaid interventional proctor for Abbott, Corindus, Scitech, Boston Scientific, and Flouit, but has not received personal payments from pharmaceutical companies or device manufacturers; being part of Argonauts, an innovation facilitator; and being partially supported by agrant from The National Council for Scientific and Technological Development (CNPq) - Brazil (grant number 306677/2019-9). The other authors have no conflicts of interest to declare.
Supplementary data
To read the full content of this article, please download the PDF.
