Abstract
Based on the latest knowledge and technological advancements, it is still debatable whether a modern revascularisation approach in the setting of acute myocardial infarction (AMI), including complete revascularisation (in patients with significant non-culprit lesions) with newer-generation highly biocompatible drug-eluting stents, requires prolonged dual antiplatelet therapy (DAPT). TARGET-FIRST (ClinicalTrials.gov: NCT04753749) is a prospective, open-label, multicentre, randomised controlled study comparing short (one month) DAPT versus standard (12 months) DAPT in a population of patients with non-ST/ST-segment elevation myocardial infarction, completely revascularised at index or staged procedure (within 7 days), using Firehawk, an abluminal in-groove biodegradable polymer rapamycin-eluting stent. The study will be conducted at approximately 50 sites in Europe. After a mandatory 30-40 days of DAPT with aspirin and P2Y12 inhibitors (preferably potent P2Y12 inhibitors), patients are randomised (1:1) to 1) immediate discontinuation of DAPT followed by P2Y12 inhibitor monotherapy (experimental arm), or 2) continued DAPT with the same regimen (control arm), up until 12 months. With a final sample size of 2,246 patients, the study is powered to evaluate the primary endpoint (non-inferiority of short antiplatelet therapy in completely revascularised patients) for net adverse clinical and cerebral events. If the primary endpoint is met, the study is powered to assess the main secondary endpoint (superiority of short DAPT in terms of major or clinically relevant non-major bleeding). TARGET-FIRST is the first randomised clinical trial to investigate the optimisation of antiplatelet therapy in patients with AMI after achieving complete revascularisation with an abluminal in-groove biodegradable polymer rapamycin-eluting stent implantation.
Introduction
The introduction of drug-eluting stents (DES) profoundly changed interventional cardiology, providing superior efficacy compared with bare metal stent implantation in treating obstructive coronary artery disease12. However, the favourable antirestenotic properties of the first-generation DES, by virtue of the antiproliferative drug impregnated into the polymer as a carrier, come with unfavourable late-onset adverse hypersensitivity reactions, delayed vessel healing, and neoatherosclerosis formation, raising concerns about the safety of these devices345. In this regard, prolonged dual antiplatelet therapy (DAPT) became the mainstay of treatment after percutaneous coronary intervention (PCI) of either stable ischaemic heart disease or, especially, after primary PCI in acute coronary syndrome (ACS)678910.
In addition to device-related thrombotic events, patients presenting with non-ST/ST-segment elevation myocardial infarction (NSTEMI/STEMI) often have a burden of multivessel coronary artery disease (CAD) with angiographically significant bystander non-culprit lesions next to the culprit lesion1112. The COMPLETE trial showed a clear benefit of complete revascularisation for the composite of cardiovascular (CV) death or myocardial infarction (MI) compared with culprit-lesion-only PCI in patients presenting with STEMI and multivessel CAD13. Furthermore, the optical coherence tomography substudy showed the presence of unstable lesions in nearly half of the patients with multivessel disease (MVD)14. Currently, international guidelines uniformly recommend at least 12 months of DAPT in ACS patients, primarily with potent P2Y12 inhibitors (ticagrelor or prasugrel), except for patients with high bleeding risk (HBR)678910. A prolonged and potent DAPT strategy efficiently ameliorates the thrombotic and ischaemic risks following ACS events but inevitably predisposes patients to bleeding complications1516171819. The heightened awareness that bleeding is a strong and independent correlate of post-PCI mortality16 coupled with the development of modern, highly biocompatible DES with lower drug doses has prompted further research to reduce or modify the DAPT strategy. Previous studies with shorter DAPT durations in the setting of ACS, using clopidogrel, showed increased ischaemic event rates compared with the guideline-recommended approach20. The additional antithrombotic effect associated with the introduction of higher potency P2Y12 inhibitors, compared with clopidogrel, resulted in a further reduction in ischaemic events, albeit with a slight increase in bleeding171819. The observed difference in treatment effects between short and prolonged DAPT regimens was most pronounced in the weeks after PCI. In contrast, the bleeding risk is related to patient characteristics, with inherent long-term risks. Therefore, a strategy to shorten DAPT duration still emerges as a justified and desirable clinical approach to achieving net clinical benefits.
In this regard, during the last decade, many trials have explored de-escalating DAPT to a single antiplatelet agent at an earlier time, but concerns about a higher rate of ischaemic events have persisted, especially after PCI for ACS20212223242526. In the recent STOPDAPT-2 ACS trial in an Asian population, a short (1 to 2 months) period of clopidogrel-based DAPT followed by clopidogrel monotherapy failed to establish non-inferiority compared with 12 months of DAPT, for the net clinical hazard of CV death, MI, definite stent thrombosis (ST), stroke, or bleeding, due to the increase in CV events having a greater impact than the reduction in bleeding events25. Contrary to those findings, the TICO trial showed a modest but statistically significant reduction in the composite outcome of major bleeding and CV events at 1 year among patients with ACS treated with ticagrelor monotherapy after 3 months of DAPT, compared with those treated with ticagrelor-based 12-month DAPT23. Similar findings were observed in the ACS subgroups of GLOBAL LEADERS and TWILIGHT2122. Hence, a more potent (and reliable) P2Y12 inhibition could pave the way for abbreviated DAPT therapy in properly selected ACS patients, i.e., those with lower bleeding and thrombotic risks and complete revascularisation.
Study rationale
Considering the iterative improvements in stent platforms (with lower device failure rates), appreciation of the importance of complete revascularisation (CR) in the reduction of new ischaemic events, along with the recognition of the association between bleeding complications and mortality, there remains an unmet clinical need to identify and test a new pharmacoinvasive strategy in patients who may not need extended DAPT and to reduce the long-term DAPT-associated bleeding risk, while maintaining early antithrombotic protection following PCI for ACS. The need for further clinical research is reflected in the main objective of the TARGET-FIRST study: to investigate whether CR (in patients with significant non-culprit lesions) using an innovative stent design in AMI patients, without significant ischaemic and bleeding risks, combined with P2Y12 inhibitor monotherapy for 11 months (following an uneventful period of 30-40 days) might provide a net clinical benefit over a standardised 12-month DAPT regimen.
Firehawk stent
The Firehawk stent (MicroPort) is a third-generation balloon-expandable rapamycin-eluting cobalt-chromium stent with a strut thickness of 86 μm (Figure 1). The stent design minimises polymer burden and reduces drug concentrations in the vessel wall. A novel feature of this device is the unique set of recessed abluminal grooves facing the coronary vessel wall, which contain a poly(D,L-lactic acid) biodegradable polymer and provide a controlled and targeted release of the antiproliferative drug rapamycin into the blood vessel wall that dissolves within 9 months. It represents the lowest polymer volume and drug concentration among currently available biodegradable-polymer DES. Despite its abluminal groove design, it has a similar mechanical performance regarding underexpansion and contraction compared to traditional DES27. In the context of these conceptual advantages, the Firehawk stent has been evaluated in several clinical trials. The most recent study is the TARGET All comers (TARGET-AC), a multicentre, open-label, randomised, non-inferiority trial. It demonstrated that the Firehawk stent is non-inferior to the benchmark durable-polymer XIENCE stent (Abbott) as assessed with the primary endpoint of target lesion failure at 12 months in an all-comers population (6.1% in the Firehawk group and 5.9% in the XIENCE group; p for non-inferiority=0.004), with a similar rate of ST and consistent outcomes up to 5 years28. The angiographic late lumen loss was similar, while the optical coherence tomography substudy at 3 months revealed near-complete strut coverage, a low rate of malapposed struts, and minimal neointimal thicknesses in both stents29.
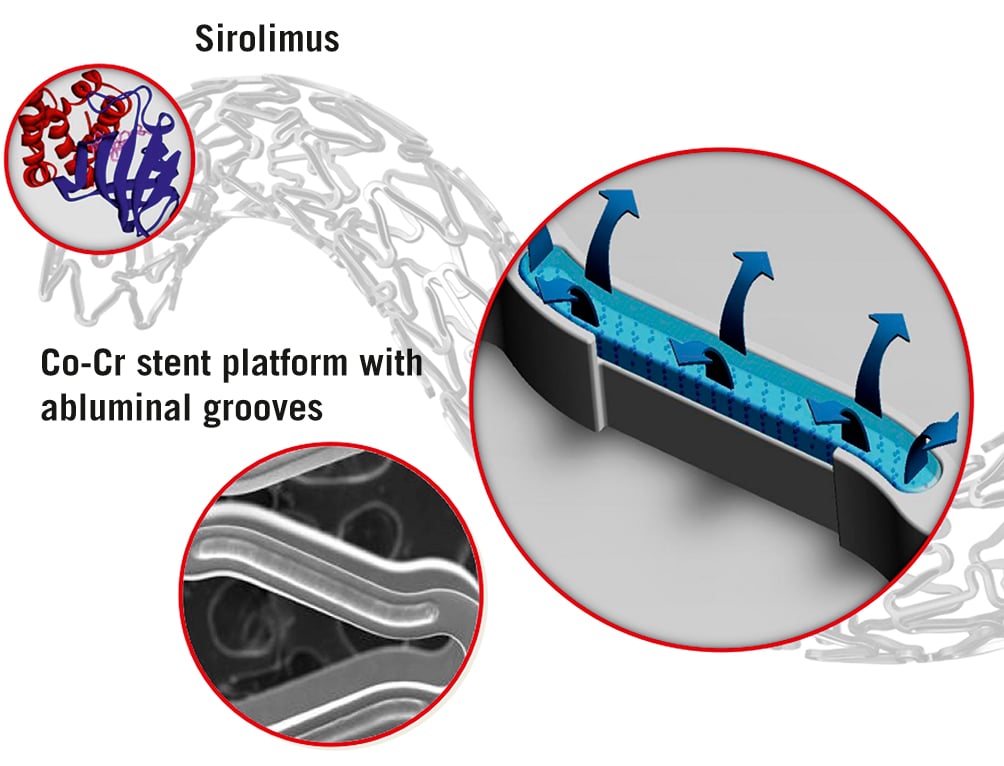
Figure 1. Firehawk stent design: cobalt-chromium (Co-Cr) stent platform with abluminal grooves (10 µm), biodegradable polymer, low dose rapamycin (0.3 µg/mm²), total strut thickness 86 µm.
Methods
Study design and population
TARGET-FIRST (Evaluation of a Modified Anti-Platelet Therapy Associated With Low-dose DES Firehawk in Acute Myocardial Infarction Patients Treated With Complete Revascularization Strategy) is a European prospective, multicentre, open-label, randomised controlled trial comparing 1 month (experimental arm) versus 12 months (control arm) of DAPT in a population of NSTEMI and STEMI patients at approximately 50 sites in Europe (Supplementary Appendix 1). A maximum of 2,246 subjects shall be enrolled until a total of 2,010 randomised patients has been attained. After a mandatory 1 month of DAPT with aspirin and P2Y12 inhibitors (preferably potent P2Y12 inhibitors), patients free of ischaemic or bleeding events are randomised (1:1) for the following 11 months to 1) discontinuation of DAPT followed by P2Y12 inhibitors only (as prescribed after index procedure) or 2) continued DAPT with the same regimen (Figure 2).
Eligible patients are those aged 18 years or more, presenting with STEMI or NSTEMI, completely revascularised at the index procedure (or staged procedure within seven days) with a Firehawk/FIREHAWK LIBERTY stent (MicroPort), without procedural complications or major cardiac events that, according to the treating physician, would preclude randomisation to the assigned antiplatelet strategy. Similarly, patients with high bleeding and ischaemic risks (due to complex PCIs) are excluded. The 7-day window for the staged procedure intends to reflect emerging standard practices and avoid an extension of the DAPT duration in the test arm. Detailed inclusion and exclusion criteria are listed in Table 1.
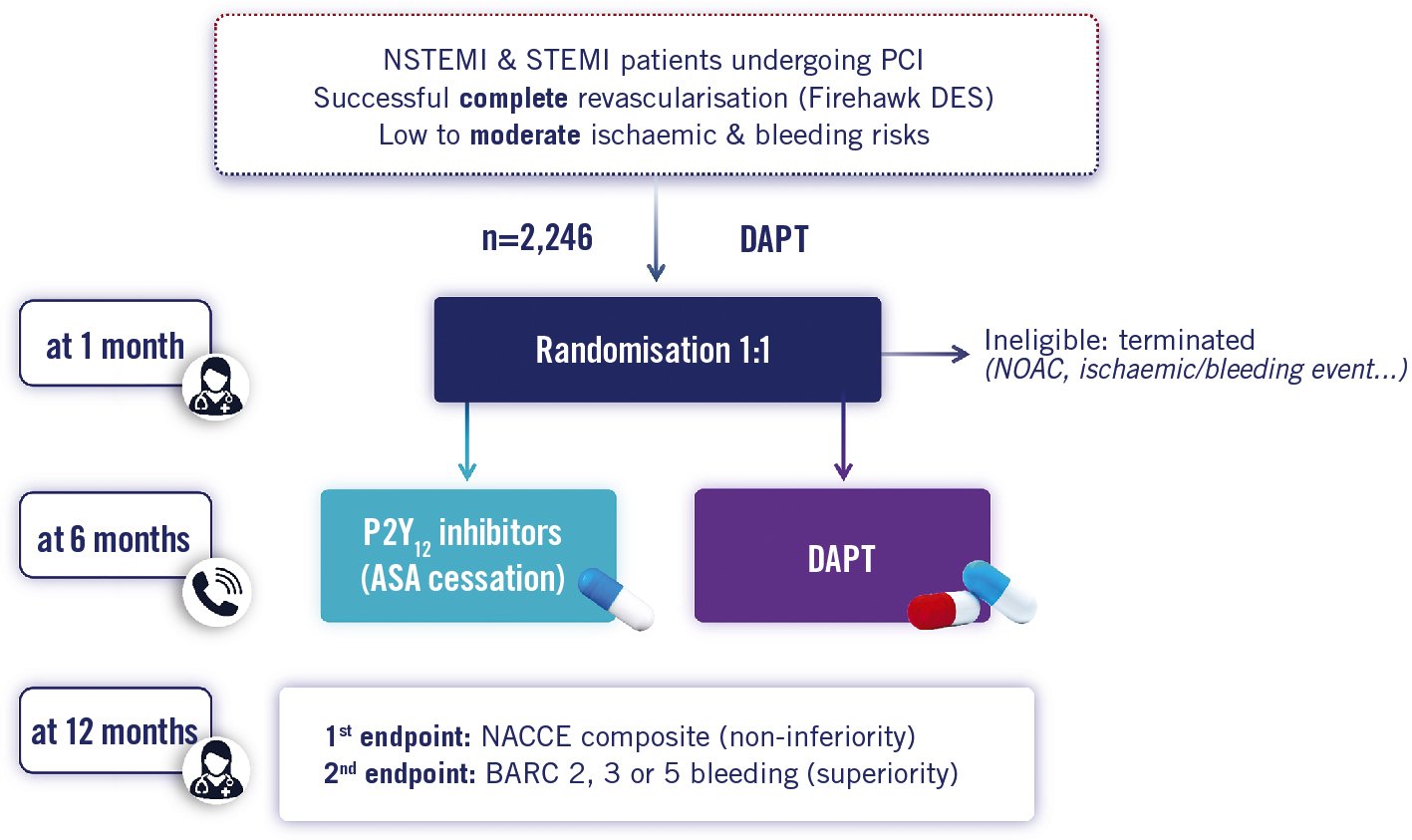
Figure 2. Study flowchart. Randomisation visit at 1 month (on site), follow-up at 6 months (telephone) and at 12 months (on site). ASA: aspirin; BARC: Bleeding Academic Research Consortium; DAPT: dual antiplatelet therapy; DES: drug-eluting stent; NACCE: net adverse cardiac and cerebral events; NOAC: new oral anticoagulant; NSTEMI: non-ST-segment elevation myocardial infarction; STEMI: ST-segment elevation myocardial infarction
Table 1. Inclusion and exclusion criteria.
| Inclusion criteria | |
|---|---|
| Clinical | Subject is ≥18 years old |
| Subject has been hospitalised for troponin-positive non-ST-elevation MI, requiring early invasive treatment (PCI), or ST-elevation MI, requiring primary PCI, and this PCI occurred within the last 7 days | |
| Subject is eligible for per protocol antiplatelet treatments | |
| Subject understands and agrees with the trial requirements and procedures, and they provide written informed consent before any trial specific tests or procedures are performed | |
| Subject is willing to comply with all protocol requirements including antiplatelet treatment strategies and follow-up visits | |
| Procedural/angiographic (related to the treatment of the [N]STEMI) | Successful revascularisation: |
| - Successful delivery and deployment of the Firehawk stent(s), with final residual stenosis of <30% (visually) for all target lesions- No occurrence of any significant event (such as MI, unplanned revascularisation, stent thrombosis, stroke, major vascular complication/bleeding). | |
| All the treated lesions: | |
| - In native coronary arteries only- In vessels with visual reference diameter ≥2.25 mm and ≤4.00 mm- Maximum 3 lesions treated*- Maximum total stent length ≤80 mm | |
| Complete revascularisation** performed when more than 1 significant lesion, during the index procedure or in staged procedure(s) occurring within 7 days from the index procedure. Physiological assessment highly recommended for lesions with stenosis between 50% and 69%. | |
| Exclusion criteria | |
| Clinical | Subjects with prior STEMI or prior PCI within 12 months before index admission |
| Prior coronary artery bypass graft (CABG) surgery | |
| Cardiogenic shock | |
| Secondary PCI | |
| Fibrinolysis | |
| Prior stent thrombosis | |
| Planned PCI, CABG, or surgery within 12 months after the enrolment | |
| Need for oral anticoagulation medications (or NOAC) | |
| Ischaemic stroke or intracerebral haemorrhage (spontaneous or traumatic) within 12 months prior to index procedure | |
| eGFR <30 mL/min/1.73 m2 or dialysis | |
| Active bleeding at time of inclusion or high risk for major bleeding | |
| History of bleeding diathesis or coagulopathy or subject refuses blood transfusions | |
| Stage B or C liver cirrhosis or active cancer within 12 months prior to index procedure (or currently receiving/scheduled to receive chemotherapy) | |
| Baseline haemoglobin <13 g/dL (12 g/dL for women) or anaemia which required transfusion in the 4 weeks prior to index procedure | |
| Moderate or severe thrombocytopenia (<100,000/L) | |
| Expected non-adherence to study protocol (such as current problems with substance abuse, severe impairment of cognitive skills, …) | |
| Estimated life expectancy ≤12 months | |
| Known hypersensitivity or contraindication to any medication used in the study or any of the study stent’s components/compounds (e.g., cobalt-chromium alloy, sirolimus, or structurally related compounds, polymer, or individual components, P2Y12 inhibitors, or aspirin). | |
| Subject participates in another interventional (device or drug) clinical trial within 12 months after the index procedure | |
| Subject is a woman who is pregnant, nursing or with known intention to procreate within 12 months after the index procedure (women of childbearing potential who are sexually active must agree to use a reliable method of contraception from the time of screening to 12 months after the index procedure). Investigator may require a pregnancy test to be performed within 7 days prior to the enrolment in women of childbearing potential) | |
| Angiographic | Any of the following: |
| - In-stent restenosis or thrombosis- Chronic total occlusion- Severe calcification- True bifurcation disease (Medina class X,X,1) and side branch diameter ≥2 mm (visual reference visual diameter) or bifurcation treated with 2 stents- Left main coronary artery lesion- Residual untreated dissection ≥C- Implantation of a non-study stent |
|
| Extent and severity of disease is such that patient is deemed to preferentially receive CABG within 1 year (based on current ESC guidelines) | |
| *in 1 to 3 vessels. **Complete revascularisation performed according to site routine practice and according to the European Society of Cardiology (ESC) guidelines. ESC: European Society of Cardiology; MI: myocardial infarction; NSTEMI: non-ST-segment elevation myocardial infarction; PCI: percutaneous coronary intervention; STEMI: ST-segment elevation myocardial infarction | |
Screening, enrolment, and randomisation phase
Patients treated with the Firehawk stent are screened for inclusion immediately after the index procedure (or staged procedure occurring within seven days of the index procedure). Patients who meet all criteria for eligibility are asked to participate in the study and are enrolled after signing an informed consent form approved by the relevant ethics committee. Consenting patients are further reassessed for eligibility during an office visit at the time of randomisation, which occurs 30-40 days after the single index or staged procedure. Patients experiencing spontaneous MI, stent thrombosis, stroke, or any revascularisation will not be randomised, and their participation in the study will be discontinued. Patients who are non-compliant or have to change prescribed DAPT because of bleeding or need for oral anticoagulant therapy will also become ineligible. At randomisation, patients are centrally allocated in a 1:1 ratio to drop aspirin and continue with the prescribed P2Y12 inhibitor or to continue with the prescribed dual antiplatelet regimen. The randomisation sequence is computer generated and stratified according to the participating site, diabetes mellitus status, and presentation with NSTEMI or STEMI. Stratification of MVD was not retained in order to limit strata and because the expected frequency and complexity of MVD is low according to the eligibility criteria.
Antiplatelet therapy
At the index procedure, the investigator will select the P2Y12 inhibitor agent (ticagrelor 90 mg twice daily, prasugrel 10 mg once a day, or clopidogrel 75 mg once a day) based on patient characteristics and per current recommendations of the European Society of Cardiology guidelines and local practice. Potent P2Y12 inhibitors are recommended but not mandated. Once the investigator has selected the P2Y12 inhibitor, it is strongly recommended that the P2Y12 inhibitor agent is not changed at any time during the study except for an urgent, well-documented medical reason (e.g., significant ischaemic or bleeding event, significant side effect, new need for treatment of arrhythmias with oral anticoagulants, etc.).
The physician must inform the patient of the prescribed antiplatelet strategy and the importance of compliance to the prescribed treatment throughout the study. The details about antiplatelet therapy (including start and stop times of interrupted therapy and reason for interruption) and other cardiac medications are recorded at each study visit.
Follow-up
Clinical follow-up visits are scheduled at 6 months (telephone or office visit) and 12 months (office visit). During these follow-up visits, an assessment of angina status (Canadian Cardiovascular Society grading), cardiovascular drug use, compliance to the antiplatelet therapy and any adverse events are recorded. The study ends at the 12-month follow-up visit.
Discussion
Study endpoints
We hypothesised that this modern approach, combining a highly biocompatible stent, complete revascularisation and modified DAPT, might be associated with similar outcomes, or a significant net benefit, compared with the guideline-recommended 12-month DAPT. The primary endpoint is net adverse clinical and cerebral events (NACCE), defined as the composite of all-cause death, myocardial infarction, definite/probable ST, stroke, or Bleeding Academic Research Consortium (BARC) bleeding (type 3 or 5) at 11 months after randomisation. Given the strong association between bleeding and mortality risks, attempts to quantify the NACCE, incorporating safety-related bleeding events aside from ischaemic complications, may provide incremental insights into understanding the risk-benefit ratio of an evaluated treatment strategy. The main secondary, statistically powered endpoint is BARC bleeding events type 2, 3 or 5 at 11 months post-randomisation. Other secondary endpoints are clinical endpoints at 1, 6 and 12 months, as detailed in Table 2.
Table 2. Primary and secondary endpoints.
| Primary endpoint | Net adverse clinical and cerebral events (NACCE) defined as a composite of all cause death, non-fatal myocardial infarction, definite/probable stent thrombosis, stroke, or Bleeding Academic Research Consortium (BARC) type 3 or 5 bleeding at 11 months post-randomisation (12 months post-index procedure). |
| Secondary endpoint (powered) | BARC type 2, 3 or 5 bleeding events at 11 months post-randomisation |
| Secondary endpoints | Other secondary endpoints (exploratory) are clinical endpoints at 1 month, 6 months and 12 months:- All-cause death, non-fatal myocardial infarction, definite/probable stent thrombosis, or stroke- BARC 3 and 5 bleeding events- All-cause death or non-fatal myocardial infarction- Patient-oriented composite of major adverse cardiac and cerebrovascular events (MACCE), including all-cause death, myocardial infarction, definite/probable stent thrombosis, any stroke, any ischaemia driven repeat revascularisation, or BARC bleeding events (type 2, 3, or 5)- Device-oriented composite endpoint of target lesion failure (cardiac death, target vessel-related myocardial infarction, target lesion ischaemia-driven revascularisation)- MACE (cardiovascular death, myocardial infarction, ischaemia-driven revascularisation)- Definite or probable stent thrombosis- BARC 3 events- BARC 5 events- BARC 2 events- Cardiovascular death- Cardiac death- Non-cardiac death- Myocardial infarction- Cardiac death or non-fatal myocardial infarction- Cardiac death, myocardial infarction, or definite/probable stent thrombosis- Cardiovascular death, myocardial infarction, definite/probable stent thrombosis, or ischaemic stroke- Ischaemic stroke- Haemorrhagic stroke- Ischaemia-driven target lesion revascularisation- Ischaemia-driven target vessel revascularisation- Cardiovascular death, myocardial infarction, or ischaemic stroke and each of the components of the primary and main secondary endpoints |
Statistical considerations and sample size calculation
The study holds two tested hypotheses that will be tested sequentially 1) primary endpoint (non-inferiority of NACCE) and 2) main secondary endpoint (superiority of BARC bleeding type 2, 3 or 5) (i.e., the main secondary endpoint will be tested if the primary endpoint is successful), in order not to inflate the alpha risk, which is fixed at 2.5% for the study. The study will be declared a success if the primary endpoint is met.
The expected rate of events (3.5% in the control group and 2.5% in the experimental group) is estimated based on the results of the SMART-CHOICE, GLOBAL LEADERS, TWILIGHT, TICO, and STOPDAPT-2 trials2021222325. The analysis aims to prove that the experimental strategy is not worse, with regards to the incidence of NACCE, by more than 1.25% compared to the standard of care (control). To provide a power of at least 80% for each tested endpoint, a sample size of 1,908 subjects (954 in each randomisation group) is needed for a non-inferiority margin of 1.25% (relative risk of 35.7%), with a 1-sided significance level (alpha) of 2.5% (Farrington-Manning method). The secondary endpoint should prove that the experimental strategy is better than the standard of care (control) for the incidence of clinically relevant non-major and major bleeding (BARC type 2, 3 or 5). For the secondary endpoint, superiority testing for the expected event rate in the experimental and control groups of 3.3% and 6.0% (the relative treatment effect of 45%), respectively, using a 2-sided 5% alpha level and with 1,908 patients, has a power of 82% (Cox’s proportional hazards model). It is deemed that 5% of the randomised subjects will be lost to follow-up and that approximately 10.5% of patients will not be randomised24. Therefore, a maximum number of 2,246 subjects should be enrolled.
The primary endpoint will be assessed in the intention-to-treat population with the Com-Nougue method, which uses Kaplan-Meier failure rate estimates for a specific timepoint and the Greenwood standard errors for these estimates. Sensitivity analyses of the primary endpoint will be performed using a per-protocol population. The secondary endpoint will be assessed with the Cox proportional hazards model that includes the randomisation group (experimental vs control) as a covariate.
The primary and main secondary endpoints will be assessed on adjudicated events according to the predefined subgroups (exploratory): age (<75, ≥75 years old), gender, NSTEMI/STEMI, mono- versus multilesions, mono- versus multivessel disease, diabetes mellitus, chronic kidney disease (estimated glomerular filtration rate < and >60 ml/min/1.73 m²), prescribed P2Y12 inhibitor, and prior myocardial infarction or percutaneous coronary intervention.
Ethical considerations, data management and personal data protection
The ethics committees and/or competent authorities, as required per applicable local/national regulations, will approve the study before any patient enrolment. The study will comply with the Declaration of Helsinki and ISO 14155. Subject data will be managed following the European Union’s General Data Protection Regulation (EU) 2016/679.
Study organisation and timelines
A steering committee is responsible for assisting the sponsor in designing the study, overseeing its scientific validity, the quality and integrity of data, and disseminating the study results through appropriate scientific presentations and publications. The multidisciplinary, independent Data Safety Monitoring Board (DSMB) monitors the safety and well-being of the participating subjects, ensures the study’s scientific integrity, and recommends actions based on potential safety issues, including study suspension or termination based on prespecified suspension criteria. An independent clinical events committee (CEC) commissioned by the European Cardiovascular Research Center (CERC, Massy, France) adjudicates all investigator-reported or otherwise triggered potential endpoint events. The CERC carries out independent data monitoring, data management and a central blinded data review, while the sponsor will perform interim safety and final statistical analyses according to the statistical analysis plan. Study enrolment began in March 2021, with 1,058 patients enrolled as of 1 December 2022. Major ischaemic events have not yet reached the predefined threshold (10 events) for the first interim safety analysis by the DSMB. Enrolment completion is expected in the fourth quarter (Q4) of 2023, and the primary endpoint results are anticipated in Q4 of 2024.
Limitations
The study population is limited to patients without significant bleeding or thrombotic risk (lower anatomical risk before PCI, particularly excluding patients with more than three lesions, more than 80 mm stent length, and bifurcation with double-stent strategy). Consequently, the MVD population has a moderately complex anatomy. Another limitation is the use of a single stent platform; thus, results cannot be generalised to other platforms.
Conclusions
TARGET-FIRST is the first trial aiming to clarify whether 1 month of dual antiplatelet therapy after STEMI or NSTEMI is safe, especially when potent P2Y12 inhibitors are used, in combination with aspirin or alone, in the context of early angiographically complete revascularisation.
Acknowledgements
The authors would like to thank all contributors: Sinisa Stojkovic, MD, PhD, for drafting the manuscript; the CEC and DSMB members; the hospitals and CERC study teams (Supplementary Appendix1-Supplementary Appendix3) and, above all, the patients who participate in this trial.
Funding
TARGET-FIRST is aclinical trial sponsored by MicroPort CRM (SORIN CRM SAS).
Conflict of interest statement
G.Tarantini has received consulting fees, payment or honoraria for lectures, presentations, speakers’ bureaus, manuscript writing or educational events from Abbott, Abiomed, Boston Scientific, Medtronic, Edwards Lifesciences, MicroPort, and GADA.P.C.Smits received personal fees as asteering member of the TARGET-FIRST study from MicroPort; consulting fees from Arena Pharmaceuticals Inc and AstraZeneca; payment or honoraria for lectures, presentations or speakers’ bureaus from Abiomed, Terumo, and Sinomed; apersonal fee for DSMB participation from Amsterdam University Hospital and Cardialysis; and institutional research grants from Dutch Heart Registration, MicroPort, SMT, Abbott Vascular, and Daiichi Sankyo. T.Lhermusier received consulting fees, payment or honoraria for lectures, presentations, speakers’ bureaus, manuscript writing or educational events from Medtronic, Abbott, Boston Scientific, and Novartis. B.Honton received consultancy fees and payment for expert testimony from Shockwave; payment or honoraria for lectures, presentations, speakers’ bureaus, manuscript writing or educational events from Medtronic, Terumo, and Shockwave; personal fees for participation on aData Safety Monitoring Board or advisory board for Terumo and MicroPort; and is an EAPCI Fellowship Grant member. G.Rangé received payment or honoraria for lectures, presentations, speakers’ bureaus, manuscript writing or educational events from MicroPort, Amgen, Sanofi, and Abbott. G.Lemesle received payment or honoraria for lectures, presentations, speakers’ bureaus, manuscript writing or educational events from Alnylam, Amgen, AstraZeneca, Bayer, Bristol-Myers Squibb, Boehringer Ingelheim, Daiichi Sankyo, Lilly, MSD, Mylan, Novartis, Novo Nordisk, Pfizer, Sanofi, and Servier. M.Godin received payment or honoraria for lectures, presentations, speakers’ bureau, manuscript writing or educational events from Medtronic and Terumo; he participated on aData Safety Monitoring Board or advisory board for Terumo and MicroPort Europe. P.Motreff received consulting fees from Terumo, Abbott, and Boston Scientific. T.Cuisset received consulting fees, payment or honoraria for lectures, presentations, speakers’ bureaus, manuscript writing or educational events, as well as support for attending meetings and/or travel, from Abbott, Medtronic, Boston Scientific, Edwards Lifesciences, and Terumo; they report participation on aData Safety Monitoring Board or advisory board at CERC and stock options in CERC.Y.Poezevara is an employee of MicroPort. D.Bouchez received consulting fees from MicroPort CRM.G.Cayla received consulting fees from Edwards Lifesciences, Medtronic, and MicroPort CRM; and payment or honoraria for lectures, presentations, speakers’ bureaus, manuscript writing or educational events from Amgen, AstraZeneca, Abbott, Bayer, Biotronik, Bristol-Myers Squibb, Edwards Lifesciences, MicroPort, Medtronic, Pfizer, and Sanofi. The other authors have no conflicts of interest to declare.
Supplementary data
To read the full content of this article, please download the PDF.
