
Abstract
Aims: We aimed to evaluate the safety and performance of a magnesium-based sirolimus-eluting metal scaffold at three-year follow-up to assess vessel response two years beyond scaffold resorption.
Methods and results: BIOSOLVE-II is an international, multicentre first-in-man study, including 123 patients with de novo lesions. Predilatation was mandatory and post-dilatation was left to the discretion of the investigators. Dual antiplatelet therapy was recommended for six months. At three years, 91.1% of patients were angina-free and 8.0% were on dual antiplatelet therapy. The target lesion failure rate was 6.8% (n=8: two cardiac deaths, one target vessel myocardial infarction and five target lesion revascularisations). No probable or definite scaffold thrombosis was observed. Imaging follow-up was voluntary and serial angiographic assessment at 6, 12, and 36 months was available in 25 patients. In these, a slight increase in in-segment and in-scaffold late lumen loss and diameter stenosis was observed between 12 and 36 months (by 0.11±0.28 mm and 0.13±0.30 mm for late lumen loss, and by 3.8±10.1% and 4.1±10.2% for diameter stenosis).
Conclusions: Two years beyond the resorption period of a sirolimus-eluting bioresorbable metal scaffold built from a proprietary magnesium alloy, complication rates remained low. In the patients with serial angiographic assessment, late lumen loss and diameter stenosis did not increase substantially beyond the resorption period.
Introduction
Bioresorbable scaffolds have been developed to overcome the limitations of permanent drug-eluting stents (DES); however, initial enthusiasm was dashed after studies raised concerns about the safety and efficacy of the polymeric Absorb™ scaffold (Abbott Vascular, Santa Clara, CA, USA) prior to its bioresorption, in particular elevated scaffold thrombosis and myocardial infarction rates1,2,3. Attempts to mitigate these risks resulted in modified implantation techniques and prolongation of dual antiplatelet therapy (DAPT), but currently it is unclear to what extent these risk mitigation measures will impact on outcomes3.
In ex vivo porcine carotid jugular arteriovenous shunt models, magnesium-based scaffolds have shown reduced thrombogenicity compared to the Absorb scaffold and to a stainless steel stent, suggesting that the magnesium-based metal scaffold may have inherent properties that reduce thrombogenicity4,5. No scaffold thrombosis has been reported for the second-generation drug-eluting absorbable magnesium scaffold (DREAMS 2G, commercial name Magmaris®; Biotronik AG, Bülach, Switzerland) in 184 patients studied up to 24 months, which is beyond its degradation time of approximately 12 months6,7,8. We now report three-year outcomes of BIOSOLVE-II (BIOTRONIK – Safety and Clinical PerFormance of the Drug Eluting Absorbable Metal Scaffold in the Treatment of Subjects with de NOvo Lesions in NatiVE Coronary Arteries) to assess vessel response up to two years post resorption.
Methods
STUDY DESIGN AND POPULATION
Study methods have been described in detail previously6,9. In brief, BIOSOLVE-II is an international, multicentre, first-in-man study assessing the safety and performance of the DREAMS 2G scaffold (Magmaris) in 123 patients enrolled in 13 centres worldwide. Patients with a maximum of two de novo lesions with a reference vessel diameter of 2.2–3.7 mm, vessel length ≤21 mm and stable or unstable angina or documented silent ischaemia could be enrolled. Main exclusion criteria were myocardial infarction within 72 hours prior to the index procedure, unprotected left main disease, three-vessel coronary artery disease, and heavily calcified lesions. The full list of inclusion and exclusion criteria is available at ClinicalTrials.gov: NCT01960504. In a subgroup of 30 consecutive patients, intravascular ultrasound (IVUS) and optical coherence tomography (OCT) was conducted pre and post procedure and at six months. Angiographic follow-up was scheduled at six months and, if the subject consented, at one and three years. If a reintervention was performed within three to six months post procedure or within the time window of the three-year follow-up, the angiographic assessment prior to the intervention was used for analysis. Lesions with reinterventions were then precluded from further imaging follow-up assessment. Additional imaging assessments outside the protocol had to be documented and were evaluated by the core laboratory. Follow-up was scheduled for up to three years.
The endpoints at three years were (a) target lesion failure (TLF), defined as a composite of cardiac death, target vessel myocardial infarction, coronary artery bypass grafting and clinically driven target lesion revascularisation (CD-TLR), and (b) stent thrombosis. Cardiac death, CD-TLR and scaffold thrombosis were defined according to the Academic Research Consortium guidelines, periprocedural myocardial infarction according to SCAI definitions and non-periprocedural myocardial infarction according to the extended historical definition10,11,12.
The study was performed according to the Declaration of Helsinki and ISO14155, was approved by the ethics committees and competent authorities, and all patients provided consent. Monitoring encompassed 100% source document verification, all events were adjudicated by a clinical events committee, and all images were assessed by a core laboratory.
DEVICE AND PROCEDURE
The Magmaris is a magnesium-based scaffold coated with bioresorbable poly-L-lactide acid which incorporates sirolimus, the same drug-polymer combination that is used for the Orsiro stent (Biotronik)13. During degradation, the magnesium alloy is first converted to hydrated magnesium oxide and, in a second phase, the magnesium oxide is converted to magnesium phosphate which is consecutively replaced by amorphous calcium phosphate. At one year, the degradation is almost fully complete (95%)9,14. The Magmaris has a strut thickness of 150 µm, a strut width of 150 µm, and was available in sizes 2.5×20 mm, 3.0×20 mm and 3.5×25 mm during the course of the study.
Predilatation was mandatory. The predilatation balloon ought to be not more than 0.5 mm smaller than the reference vessel diameter and should not exceed the reference vessel diameter. Furthermore, it should not exceed the length of the original lesion. Post-dilatation was left to the discretion of the investigator; however, the diameter of the post-dilatation balloon should not exceed the selected diameter of the scaffold by more than 0.5 mm. DAPT was recommended for at least six months post procedure.
STATISTICAL ANALYSIS
The sample size had been calculated for the primary endpoint, late lumen loss (LLL) at six months9. The analysis was based on the intention-to-treat population, defined as patients in whom the scaffold entered the guide catheter. Patients not receiving the scaffold counted towards device and procedure success only. For continuous data, means, standard deviations and 95% confidence intervals (CI) were calculated, as appropriate. For categorical data, absolute and relative frequencies with 95% CI for proportions were calculated.
For clinical outcomes, the denominator included patients with a respective event and/or follow-up assessment. Student’s t-test, Wilcoxon sign test and Fisher’s exact test were applied for comparison; p-values of <0.05 represent significance. A post hoc analysis was performed to compare outcomes beyond 12 months with those of an unaffected vessel without stenosis to estimate general disease progression. For that purpose, the normalised LLL (LLL divided by reference vessel diameter) in target vessels and similar non-target vessels with comparable dimensions were compared. All statistical analyses were performed with SAS version 9.4 (SAS Institute, Cary, NC, USA).
Results
Outcomes up to two years have been described previously6,9,15. In brief, lesions were 12.6±4.5 mm long with a diameter of 2.68±0.40 mm. Type A, B1, B2 and C lesions were present in 0.8%, 55.7%, 41.8%, and 1.6%, respectively. Post-dilatation was performed in 61.2% of lesions with a mean pressure of 18.0±4.5 atm (95% CI: 17.1-19.0). In two patients, the scaffold could not be implanted; these patients were not included in the angiographic and clinical endpoint analysis according to the clinical investigation plan.
At six months, 90.6% (106/117) of patients were on DAPT, eight patients received anticoagulation therapy for atrial fibrillation, and three patients were on acetylsalicylic acid only. At 12 months, 44.4% (52/117) of patients were on DAPT, and at three years 8.0% (9/112). The ischaemic status at three years was assessed in 112 patients, of whom 91.1% (n=102) were symptom-free and 8.9% (n=10) had stable angina.
Clinical data at three years were available for 117/121 patients (97%; two patient visits were missed and two patients withdrew consent). One additional TLF occurred between two and three years, leading to a three-year TLF rate of 6.8% (n=8 [95% CI: 3.0-13.0]) (Table 1). Clinically driven TLR occurred in five patients (4.3% [95% CI: 1.4-9.7]) (Supplementary Table 1). Except for one periprocedural infarction (0.9% [95% CI: 0.0-4.7]), no target vessel myocardial infarction was observed and there was no definite or probable scaffold thrombosis. Of the five deaths, two were cardiac (one unwitnessed death on day 134 and one on day 395); the others were cancer, pulmonary infection, and intracerebral haemorrhage.
Voluntary angiographic assessment at 36 months was performed in 48 patients. There was no significant difference in baseline and procedural characteristics between patients with 36-month angiography and those without, except that patients with 36-month angiography had smaller minimal lumen diameters at baseline and a lower maximum pressure at implantation (Supplementary Table 2). LLL at 36 months was 0.43±0.40 mm in-segment and 0.54±0.38 mm in-scaffold, and diameter stenosis was 28.6±11.6% (range 0.8 to 58.3) and 26.7±11.9% (range 0.8 to 60.0), respectively.
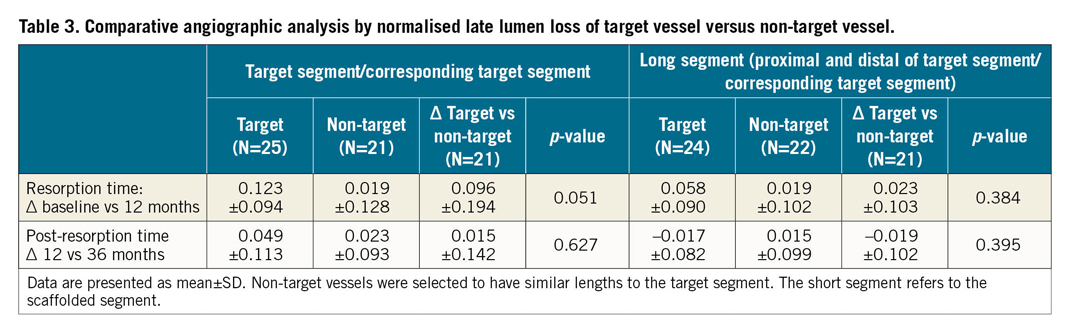
Serial angiographic assessments at 6 and 36 months are available for 47 patients, and pairs for 6, 12, and 36 months are available for 25 patients (Figure 1, Supplementary Table 3). The increase in LLL between one and three years was 0.1±0.28 mm in-segment (range –0.40 to 0.66, p=0.060) and 0.13±0.30 mm in-scaffold (range –0.33 to 0.75, p=0.042). Diameter stenosis increased by 3.8±10.1% in-segment (range –11.5 to 24.1%, p=0.072) and 4.1±10.2% in-scaffold (range –11.5 to 28.5%, p=0.054). Serial OCT (n=12) and IVUS (n=8) assessments are shown in Figure 2, Table 2, Supplementary Table 2, and Supplementary Table 4. No intraluminal mass or prolapse was detected by OCT.
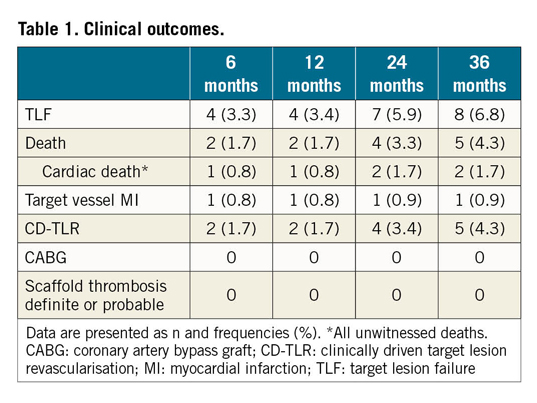
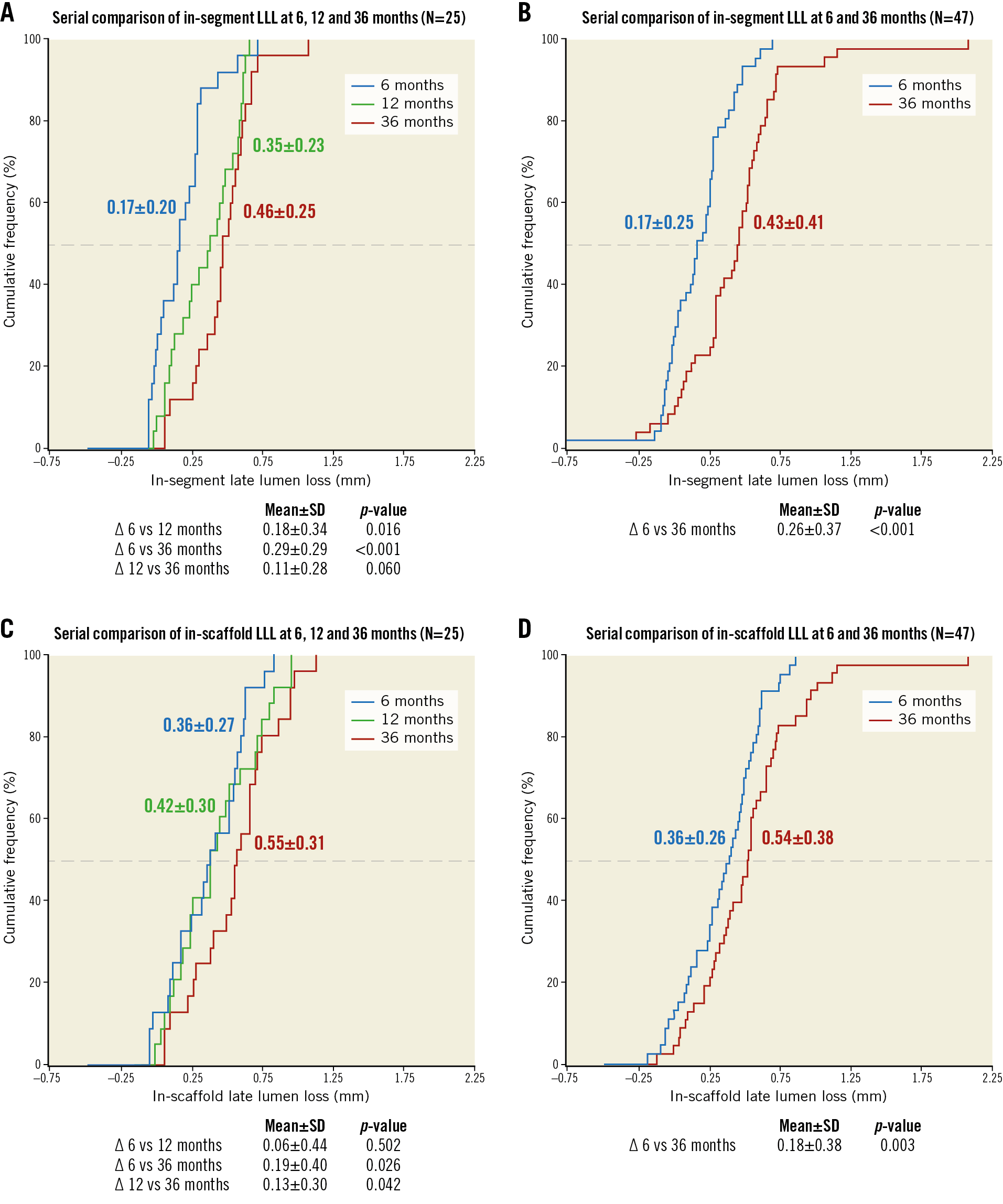
Figure 1. Cumulative frequency curves for in-segment and in-scaffold late lumen loss up to 36 months. A) & B) Serial in-segment late lumen loss (LLL) at 6, 12, and 36 months in 25 patients and serial LLL at 6 and 36 months in 47 patients. C) & D) Outcomes for in-scaffold LLL. Note: angiographic follow-up was mandatory at 6 months and voluntary at 12 and 36 months.
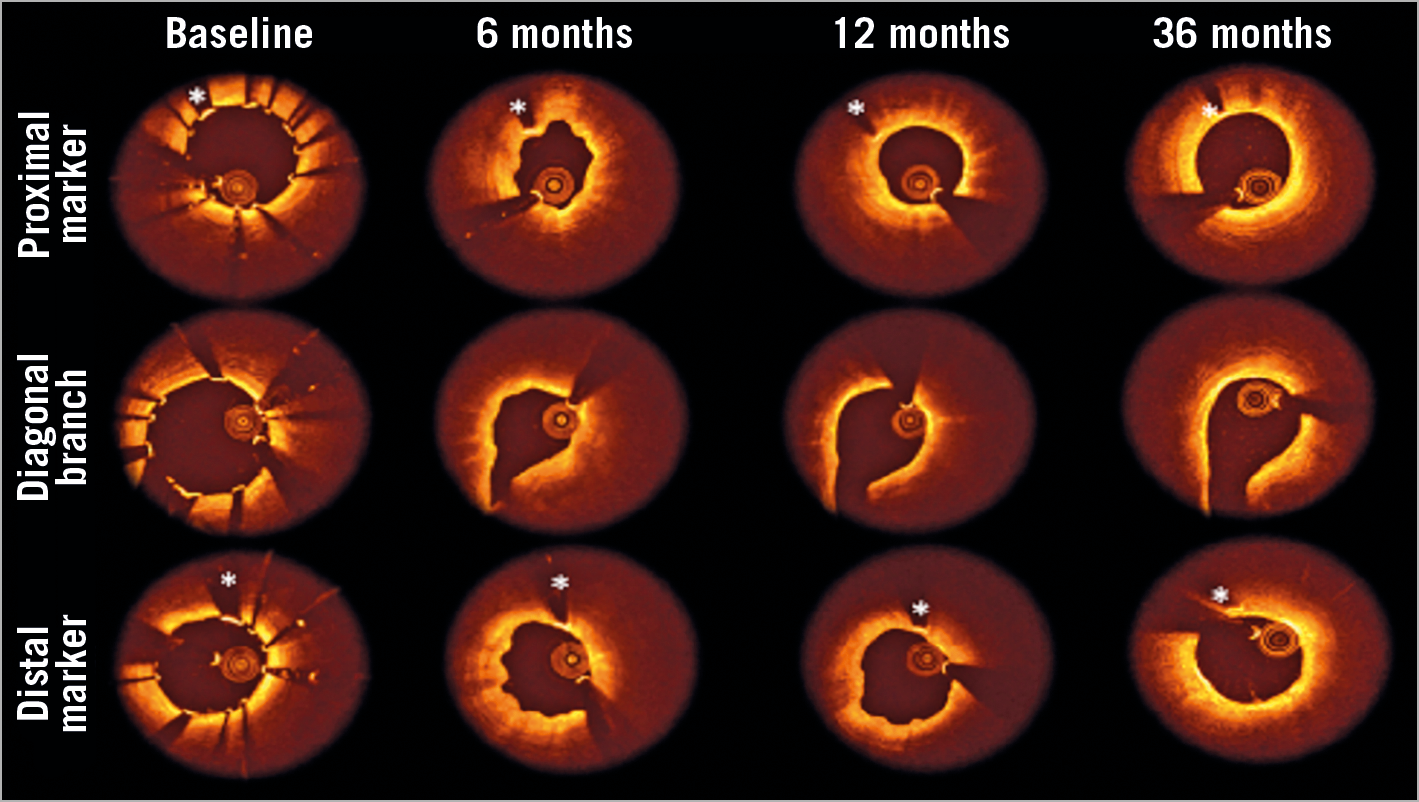
Figure 2. Serial OCT analysis from post procedure to 6, 12 and 36 months. A representative example of serial OCT assessment proximal, at the origin of the diagonal branch and at the distal end of the scaffold. The frames demonstrate the degradation of the scaffold over time with struts covering the side branch already resorbed at six months. The lumen area slightly decreased during the 12-month degradation of the scaffold. At 36 months, a slight increase in lumen area can be observed. *scaffold marker.
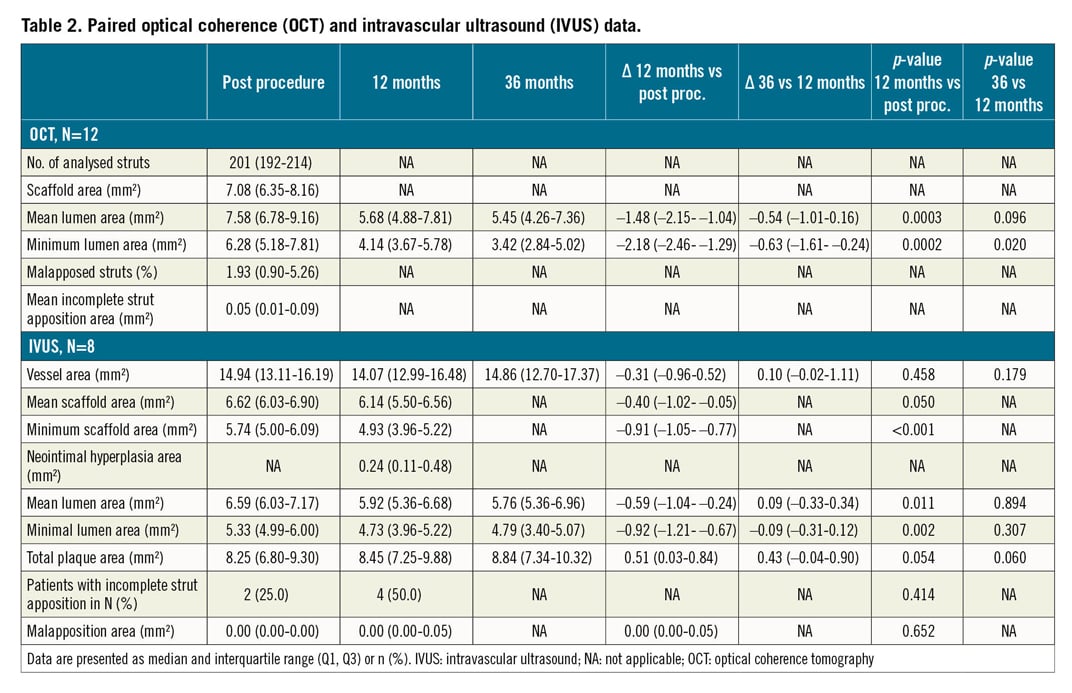
In a post hoc analysis, the normalised LLL of study scaffolded vessel segments (“target segments”) was compared to the corresponding target segments of non-target vessels of similar dimensions (Figure 3, Table 3). In the scaffolded target segments, there was a strong trend towards a higher normalised LLL compared to the corresponding target segments of non-target vessels at 12 months, while beyond 12 months the normalised LLL was not statistically different between target and non-target vessels. When analysing the “long segments” (segments proximal and distal to the target respective to the corresponding target vessel segment), there was no difference between the target compared to the non-target segments in normalised LLL at 12 months and 36 months.
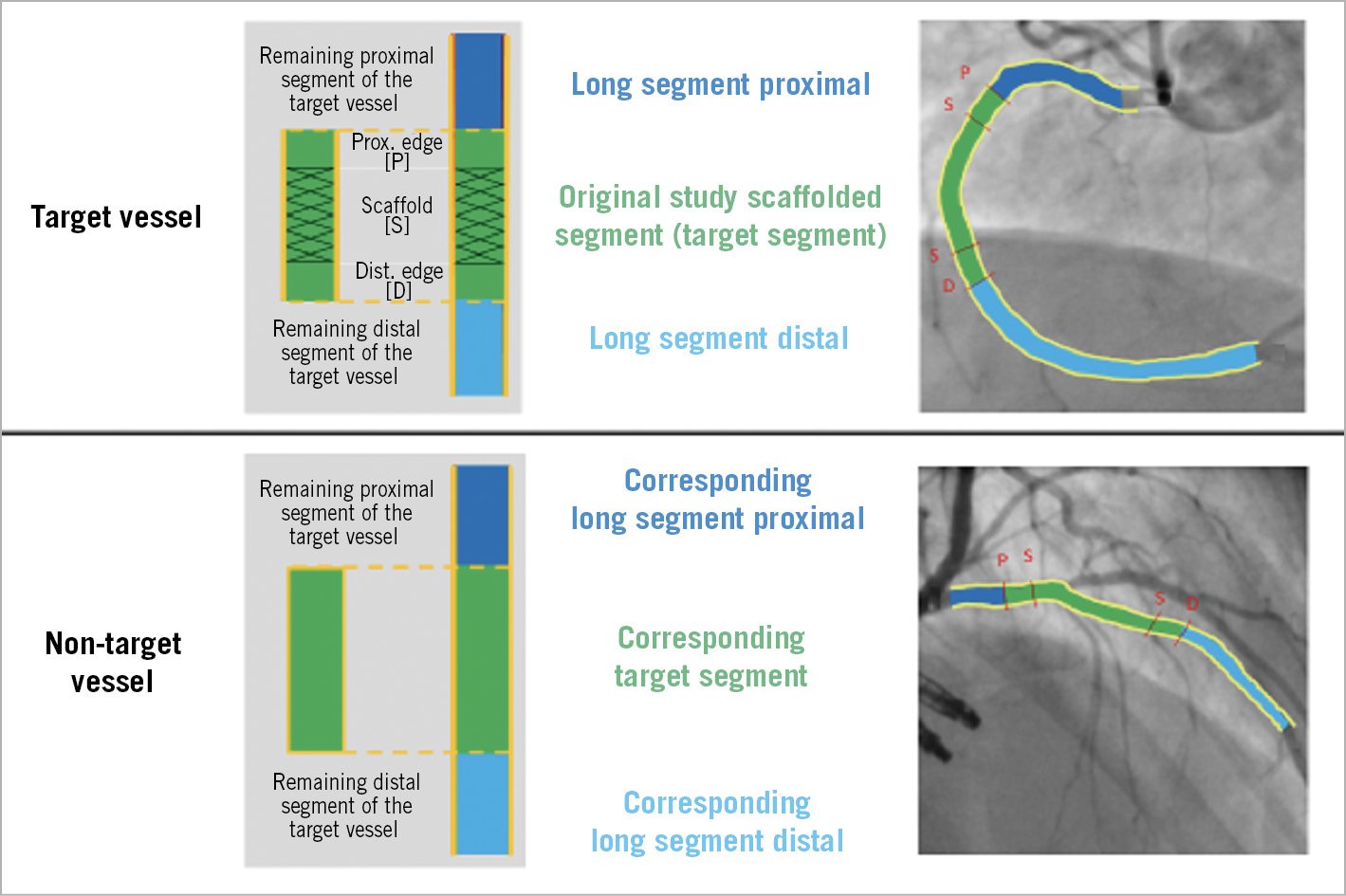
Figure 3. Comparative angiographic analysis of target vessel versus non-target vessel (without stenosis). Schematic procedure of how the attribution of the normal progression of the disease was estimated within the target and the non-target vessel.
Discussion
The main findings of BIOSOLVE-II at three years are (a) sustained low TLF and TLR rates, (b) absence of definite or probable scaffold thrombosis, and (c) stable lumen dimensions between 12 and 36 months.
TLF and CD-TLR rates were similar to three-year outcomes of the Absorb scaffold and the everolimus-eluting XIENCE® stent (Abbott Vascular) obtained from a recent patient-data pooled meta-analysis16. In this meta-analysis, the pooled TLF rate was 11.7% for Absorb and 8.1% for XIENCE compared to 6.8% in our series; the CD-TLR rate was 6.6% for Absorb and 4.4% for XIENCE compared to 4.3% in our series. The three-year CD-TLR rate of Absorb in the ABSORB cohort B trial was 7.0%17. To the best of our knowledge, no three-year outcomes of other CE-marked scaffolds have been published to date. However, 24-month outcomes of the DESolve® polymeric scaffold (Elixir Medical Corporation, Sunnyvale, CA, USA) were similar to 36-month outcomes in our series (7.4% TLF and 4.1% TLR)18.
Consistent with previous reports of BIOSOLVE-I, II and III studies6,9,19, no definite or probable scaffold thrombosis was observed at 36 months compared to 0% for ABSORB cohort B, 2.4% for Absorb and 0.6% for XIENCE in a meta-analysis of randomised trials16,17, and 0.8% for DESolve at 24 months18. These outcomes were obtained even though only 44.4% of patients were on DAPT at 12 months and 8.0% at 36 months. In contrast, prolonged DAPT covering the resorption period is recommended for Absorb1,16; in ABSORB II, DAPT use at three years was 31% for Absorb and 30% for XIENCE20, and, in ABSORB III, DAPT use was 55.8% and 53.5%, respectively21. There are multiple explanations for these outcomes: first, the absorption time of Magmaris/DREAMS 2G is substantially shorter than for Absorb (approximately 12 months versus approximately three years13,17); second, Magmaris/DREAMS 2G seems to have fewer issues such as malapposition, disintegration or discontinuities6,16; and third, the Magmaris scaffold itself is associated with reduced thrombogenicity as shown in porcine arteriovenous shunt models in which Magmaris exhibited less thrombogenicity and inflammatory cell deposition compared to the Absorb scaffold and an equivalent 316L stainless steel stent4,5.
Published three-year angiographic outcomes of contemporary stents in similar populations to BIOSOLVE-II are rare but, if reported, LLL and diameter stenosis were superior to our series (except for a similar in-scaffold diameter stenosis of Absorb). In our series, in-scaffold LLL at 36 months was 0.54±0.38 mm compared to 0.29±0.43 mm in ABSORB cohort B17, 0.37±0.45 mm for Absorb and 0.25±0.25 mm for XIENCE in ABSORB II20, and 0.25±0.37 mm for the biodegradable polymer sirolimus-eluting Firehawk® stent (MicroPort Medical, Shanghai, China) and 0.26±0.19 mm for the durable polymer everolimus-eluting XIENCE V stent in the TARGET I trial22. In-scaffold diameter stenosis was 26.7±11.9% in BIOSOLVE-II compared to 23.2±14.9% for ABSORB cohort B17, 25.8±17.3% for Absorb and 15.7±8.3% for XIENCE in ABSORB II20, and 13.2±11.0% and 12.6±6.8% in TARGET I for Firehawk and XIENCE V stents22.
In contrast to outcomes of BIOSOLVE-I with the precursor product DREAMS first generation19, no decrease in mean LLL was observed in BIOSOLVE-II up to three years. This might be a play of chance due to the low number of serial imaging follow-ups in BIOSOLVE-I and BIOSOLVE-II or might be related to different confounding patient and lesion characteristics. However, the lumen size was reported to be preserved between six and 12 months15. Most importantly, the LLL between 12- and 36-month follow-up remained stable and was similar between target and non-target segments. Therefore, the change in LLL may be attributed to the overall disease progression rather than very late effects of DREAMS 2G beyond its resorption time. Notably, in serial assessments of permanent DES, it seems that there is a more pronounced increase over time. For instance, in the TARGET I trial, both contemporary stents had an LLL of only 0.05 mm at nine months, which increased by 0.20 mm and 0.21 mm to 0.25 and 0.26 mm at 36 months, respectively22, while in BIOSOLVE-II the difference between one and three years was 0.13 mm.
Limitations
BIOSOLVE-II has several limitations which have been reported previously6,9. In brief, the trial was not randomised and included patients with more simple lesions. The lack of mandated imaging follow-up beyond six months and the associated low patient number with follow-up assessments is the main limitation of the study; therefore, imaging outcomes should be interpreted with caution. As the imaging follow-up beyond six months was voluntary, there might be a potential selection bias, even though the comparison of baseline and procedural parameters did not reveal any relevant differences among the groups. Furthermore, the comparison of disease progression in target versus non-target vessels should be interpreted considering that it was a post hoc analysis and disease progression may vary in different vessels. Even though these data present outcomes two years beyond the resorption time, further follow-up would be interesting to assess how the disease progression will evolve over time. Furthermore, registry data are awaited to report outcomes in a larger patient population.
Conclusions
In BIOSOLVE-II, the drug-eluting metal magnesium-based scaffold DREAMS 2G/Magmaris showed favourable safety outcomes at three years, with low TLF and TLR rates and absence of definite or probable scaffold thrombosis in a patient cohort with common risk characteristics for a first-in-man trial. There was no substantial increase in LLL from one to three years.
|
Impact on daily practice Three-year outcomes of BIOSOLVE-II provide additional assurance on the long-term outcomes of DREAMS 2G (Magmaris) with clinical event rates comparable to contemporary permanent DES; the absence of definite and probable scaffold thrombosis two years beyond the resorption period is encouraging. While lumen size was preserved beyond the resorption period, overall angiographic outcomes are somewhat disappointing. |
Acknowledgements
We thank Beatrix Doerr for her expert medical writing assistance, reimbursed by Biotronik AG.
Funding
This study was funded by Biotronik AG, Bülach, Switzerland.
Conflict of interest statement
M. Haude reports study grants and personal fees from Biotronik, Abbott Vascular, Cardiac Dimensions, and Philips. E.H. Christiansen reports grants from Biotronik. R. Toelg reports personal fees from Biotronik and Abbott Vascular. P.A. Lemos reports grants from Biotronik. C. von Birgelen reports grants from Abbott Vascular, Biotronik, Boston Scientific, and Medtronic. W. Wijns reports institutional grants from Abbott Vascular, Biotronik, MicroPort and Terumo, is a co-founder of Argonauts Partners and Celyad, and receives speaker fees from Biotronik and MicroPort. F-J. Neumann reports grants from Biotronik, Edwards Lifesciences, Medtronic, Bayer Healthcare, Abbott Vascular, Novartis, Pfizer, GlaxoSmithKline, and Boston Scientific. E. Eeckhout reports speaker honoraria and research grants from Biotronik. H.M. Garcia-Garcia and R. Waksman report that MedStar was the core laboratory of the study, and R. Waksman reports grants and personal fees from Abbott Vascular, AstraZeneca, Biosensors, Biotronik, Boston Scientific, Chiesi, personal fees from Amgen, Corindus, Lifetech Medical, Medtronic, Philips Volcano, Pi-Cardia Ltd and CardioSet, is an investor of MedAlliance, and has received grants from Edwards Lifesciences. The other author has no conflicts of interest to declare.
