
Abstract
Bioresorbable scaffolds represent an exciting milestone in the development of coronary stent technology with the potential to substantially improve the management of patients with coronary artery disease. In an attempt to provide first recommendations for the technology, experienced experts involved in the first-in-man studies met in Zurich on the 14 April 2016 in order to reach consensus on a responsible market introduction. This document will be updated regularly as new information from clinical trials becomes available and should be understood as a review of current data, opportunities, expectations, advice, and recommendations for future investigations.
Introduction
Bioresorbable scaffolds represent an exciting milestone in the development of coronary stent technology with the potential to substantially improve the management of patients with coronary artery disease. The technology is still in its infancy, but expectations of a fully absorbable scaffold with the potential to restore vascular reactivity are very promising1.
Prior launch of the Absorb™ (Abbott Vascular, Santa Clara, CA, USA) PLLA-based bioabsorbable scaffold in Europe for unrestricted use resulted in higher than anticipated scaffold thrombosis. Due to this, further training and adjustments to the technique were required, as well as improved patient and lesion selection.
Magnesium scaffolds exhibit different mechanical properties compared to PLLA scaffolds in terms of deliverability, radial force and degradation process. However, due to the novelty of the technology, recommendations have to be defined on the type of patients who would benefit most from the implantation of magnesium bioresorbable scaffolds. Additionally, targeted subgroups for further investigations are to be identified. Besides patient selection, procedure optimisation – particularly lesion preparation and sizing – has to be discussed.
In an attempt to provide first recommendations for this technology, experienced experts involved in the first-in-man studies met in Zurich on 14 April 2016 in order to reach consensus on a responsible market introduction.
The panel recognised that for the time being clinical data are sparse and extra caution should be used in this task. This document will be updated regularly as new information from clinical trials becomes available and should be understood as a review of current data, opportunities, expectations, advice and recommendations for future investigations. It is not intended as a definitive statement questioning official guidelines. As in any procedure, the physician has to take the final decision based on the patient’s individual history, comorbidities and lesion characteristics.
Magmaris: short overview of the technology and device
Magmaris (Biotronik AG, Bülach, Switzerland) is a balloon-expandable sirolimus-eluting bioresorbable scaffold on a rapid-exchange delivery system (Table 1, Table 2) which received CE approval in Europe in June 2016. The scaffold backbone is made from an absorbable magnesium alloy with two permanent X-ray tantalum markers at the distal and proximal scaffold end. About 95% of the magnesium is resorbed within 12 months. The resorption of the magnesium backbone takes place in two steps: first, the magnesium alloy reacts with water to create magnesium hydroxide; second, the magnesium hydroxide is slowly converted to an amorphous calcium phosphate phase with a high water content. The surface of the scaffold backbone is completely coated with bioresorbable poly-L-lactic acid which incorporates sirolimus, a successfully proven coating used in the Orsiro stent (Biotronik AG). The sirolimus load is 1.4 µg/mm2 of scaffold surface. Poly-L-lactic acid is highly biocompatible and undergoes self-catalysed hydrolytic degeneration to lactic acid, which eventually metabolises and is transformed into CO2 and H2O 2.
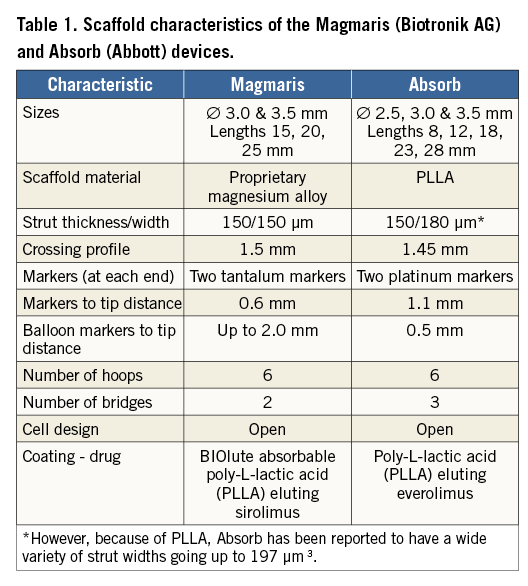
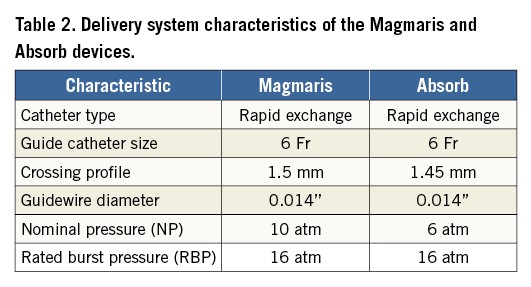
Due to the metallic properties of the magnesium alloy, Magmaris shows improved deliverability compared to Absorb4. Indeed, track and push were tested in a bench test comparing six Magmaris against six Absorb GT1 devices using a pooled variance t-test for independent samples to calculate the p-values. Magmaris showed a 29% reduction in the peak force needed to track through a tortuous vessel (Magmaris 1.70±0.21 N vs. Absorb GT1 2.40±0.21 N, p<0.001)4 and a 34% increase in the force transmitted from the hub to the tip (Magmaris 45.4% vs. Absorb GT1 33.8%, p=NS)3. Results are summarised in Table 3.
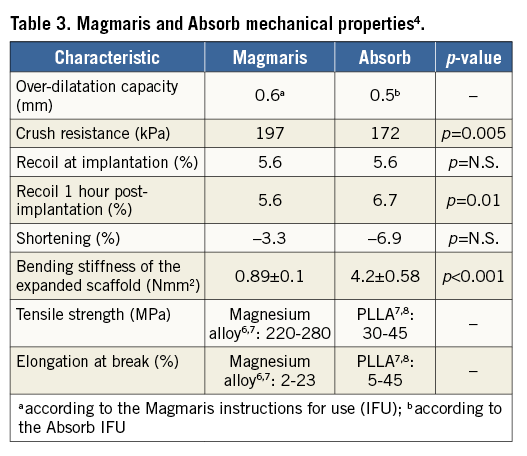
Other stent materials, such as cobalt chromium and stainless steel, have a high tensile strength (>1,000 MPa and 670 MPa, respectively)5 and elongation at break (>50% and 48%, respectively)5, which is to be expected considering the nature of these elements. However, being permanent metals, they remain in the patients’ arteries lifelong after implantation.
Magmaris is currently being tested in the clinical trials BIOSOLVE-II9 and BIOSOLVE-III. In summary, BIOSOLVE-II is an international, prospective, multicentre, non-randomised, first-in-man trial carried out in 13 centres. Eligible patients had stable or unstable angina or documented silent ischaemia and a maximum of two de novo lesions in two separate coronary arteries with a reference vessel diameter between 2.2 mm and 3.7 mm, a lesion length of 21 mm or less, and a stenosis of between 50% and 99% in diameter. Exclusion criteria included a left ventricular ejection fraction of less than 30%, thrombus in the target vessel, severe calcification, three-vessel disease, ostial lesion, target lesions involving a side branch of more than 2.0 mm in diameter, target lesions located in or supplied by an arterial or venous bypass graft and unsuccessful predilatation. The full list of inclusion and exclusion criteria can be accessed at ClinicalTrials.gov (number NCT01960504). One hundred and twenty-three patients were enrolled and clinical follow-up was scheduled at one, six, 12, 24, and 36 months. Patients were scheduled for angiographic follow-up at six months and a subgroup of 30 patients was scheduled for intravascular ultrasound (IVUS), optical coherence tomography (OCT), and vasomotion assessment. The primary endpoint was in-segment late lumen loss at six months. Dual antiplatelet therapy (DAPT) was prescribed for a minimum of six months for all patients according to the ESC guidelines.
At six-month follow-up, mean in-segment late lumen loss was 0.27±0.37 mm and angiographically discernible vasomotion was documented in 80% of the cases. IVUS assessment showed a preservation of the scaffold area with a low neointimal area, and OCT did not detect any intraluminal masses. Target lesion failure (TLF) occurred in 3.3% of the patients: one event (0.8%) was adjudicated as cardiac death, one patient (0.8%) had a periprocedural myocardial infarction (MI) and two patients (1.7%) needed clinically driven target lesion revascularisation (TLR).
The findings from BIOSOLVE-II show promising results in terms of both safety and efficacy for the use of Magmaris in de novo coronary lesions. The 12-month data were published in the European Heart Journal10. Briefly, the results demonstrated a continuous favourable safety profile of the device up to 12 months and stable angiographic parameters between six and 12 months.
In addition to BIOSOLVE-II, Magmaris is also being investigated in the ongoing BIOSOLVE-III and upcoming BIOSOLVE-IV trials.
BIOSOLVE-III, a registry currently enrolling, aims to investigate Magmaris in 61 patients with a maximum of two de novo lesions in two separate coronary arteries. Eligible patients had stable or unstable angina or documented silent ischaemia. Reference vessel diameters ranged between 2.7 mm and 3.8 mm by visual estimation with a maximum lesion length of 21 mm. Exclusion criteria included: evidence of myocardial infarction within 72 hours prior to PCI, a more than twofold elevation of CK level or threefold elevation of CKMB above the upper range limit within 24 hours prior to procedure, left main coronary disease, three-vessel disease at the time of procedure, thrombus in the target vessel, ostial lesions and lesions involving a side branch of more than 2.0 mm in diameter and severe calcification. The full list of inclusion and exclusion criteria can be accessed at ClinicalTrials.gov (number NCT02716220). The primary endpoint is procedure success during hospital stay defined as a final diameter stenosis of <30% measured by QCA without death, MI or TLR. An important secondary endpoint will be a mandatory angiographic follow-up at 12 months.
BIOSOLVE-IV, a post-market registry, will investigate Magmaris in 1,065 patients needing treatment of de novo native coronary artery lesions. Target lesion stenosis by visual estimation should be more than 50% with TIMI flow equal to or greater than one. Patients on dialysis or with known allergies to aspirin, clopidogrel, ticlopidine, heparin or any other anticoagulant/antiplatelet required for PCI, contrast medium, sirolimus, or similar drugs, or the scaffold materials including magnesium, yttrium, neodymium, zirconium, gadolinium, dysprosium, tantalum that cannot be adequately pre-medicated, were excluded. The primary endpoint is TLF at 12 months defined as a composite of cardiac death, target vessel MI and TLR.
Patients expected to potentially benefit or not from a scaffold
Magmaris received CE mark approval in June 2016. The device has not been used outside controlled clinical trials, and current experience is limited to fewer than 200 patients from the BIOSOLVE-I, II and III studies. Experience and results with other bioresorbable scaffolds are used to define these first recommendations.
TECHNICAL PROCEDURAL ASPECTS
The experts recommend a careful technique for vessel preparation, deployment and assessment of the Magmaris. This includes precise assessment of vessel size and lesion length. Meticulous vessel preparation is highly advised and image-guided implantation is highly recommended in the initial phase of the learning curve to optimise deployment. Image guidance could help deciding whether post-dilatation is required.
The Magmaris IFU recommend a vessel diameter between 2.7 mm and 3.2 mm for a scaffold diameter of 3.0 mm and a vessel diameter between 3.2 mm and 3.7 mm for a scaffold diameter of 3.5 mm. Moreover, upsizing of the device should be limited to 0.6 mm beyond the nominal size of the implanted scaffold. While balloon predilatation should be required for all lesions, balloon post-dilatation with a non-compliant balloon inflated with a pressure greater than 16 atmospheres and with the same nominal size as the scaffold implantation balloon or up to 0.5 mm larger is always recommended if the implant result is not controlled by intracoronary imaging with documentation of good strut apposition, less eccentricity and <20% residual stenosis.
Physicians should avoid planned overlapping. In the event that a second or a further scaffold is necessary, a second Magmaris should be considered first, positioning it scaffold to scaffold without an overlap or gap (Figure 1). If a DES is elected to be used directly adjacent to or overlapping a Magmaris, Orsiro with its ProBIO passive coating is recommended.
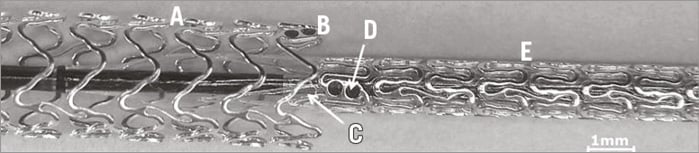
Figure 1. Optimal positioning of two adjacent Magmaris scaffolds. A) Expanded Magmaris. B) Tantalum markers of the expanded Magmaris. C) Balloon marker of the Magmaris delivery system. D) Tantalum markers of the unexpanded Magmaris. E) Unexpanded Magmaris on the delivery balloon.
The anticoagulation regimen during the procedure and the antiplatelet therapy post-procedure should be the standard of care for PCI with DES. While there are no stent thrombosis reports with Magmaris in clinical trials, the panel recognises that the current data are very limited and does not suggest any deviation from the current ESC/EAPCI guidelines of a minimum of six months post-deployment for stable patients9,10.
PATIENT SELECTION
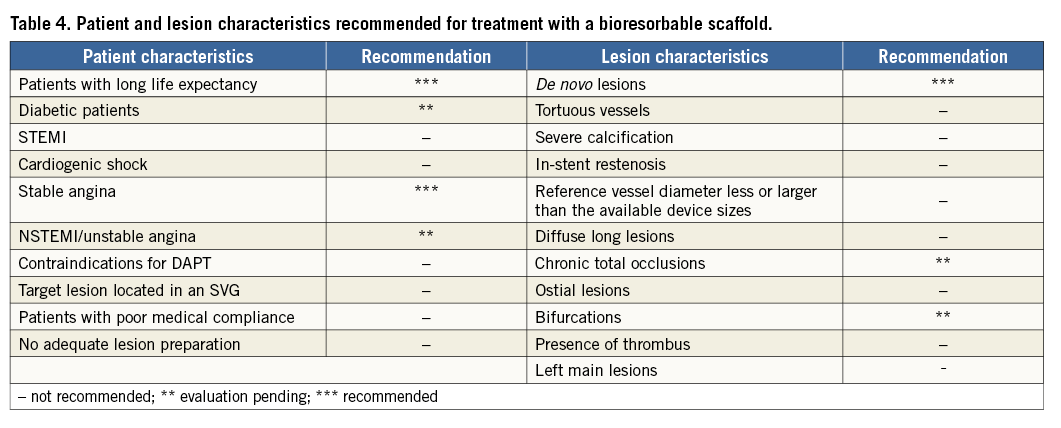
The group discussed the optimal patient population and lesion subset for the introduction of this technology. A summary of the recommendations is displayed in Table 4:
It was agreed that the potential advantages of the technology would most benefit patients with a long life expectancy. The panel agreed that gender or race should not influence the use of the device, and the device should be used initially in stable patients with discrete stable short de novo lesions. Patients with de novo lesions would benefit from a bioresorbable scaffold as vasomotion is expected to return six months after the implantation.
Lesion size and length should be carefully assessed to match the matrix of sizes and lengths available, and to prevent overlapping.
In contrast, the following patient groups are currently not within the recommendations for treatment with a bioresorbable scaffold:
PATIENTS FOR WHOM ADEQUATE LESION PREPARATION CANNOT BE OBTAINED
If full expansion of the pre-implantation balloon and a residual stenosis of less than 30% cannot be achieved, the use of Magmaris is not recommended. With respect to vessel size, if the vessel accepts a pre-implantation balloon size of at least 3.0 mm, then a 3.0 mm Magmaris can be used. As soon as a 2.5 mm sized Magmaris is available, a minimal lumen diameter of 2.5 mm or above can be treated.
PATIENTS WITH A REMAINING THROMBUS AT THE LESION SITE
Data are lacking for this subgroup and unfavourable data from the Absorb scaffold11-14 lead to the recommendation to use established stent technologies if there is a thrombus at the lesion location at the time the scaffold is intended to be implanted.
PATIENTS FOR WHOM RETURN OF VASOMOTION CANNOT BE EXPECTED
For example, saphenous vein grafts, in-stent restenoses, previous stents in the same vessel segment and highly calcified lesions are not recommended for a scaffold, because vasomotion cannot be expected to return.
PATIENTS FOR WHOM PROPER SIZING CANNOT BE ACHIEVED
QCA, IVUS and OCT should be employed to measure the vessel size and help in the choice of the proper device size at least at the beginning of the learning curve.
LEFT MAIN LESIONS
Left main lesions have a lumen diameter which is usually too large to treat with the current available scaffold sizes. To avoid malapposition or fractures caused by an excessive post-dilatation of the scaffold, left main lesions should not be treated with current bioresorbable devices.
DAPT CONTRAINDICATIONS
DAPT recommendation of six months post-deployment is based on protocol-mandated DAPT in the BIOSOLVE-II study as well as the currently ESC/EAPCI guidelines for stable patients9,10. Patients who cannot comply with current DAPT guidelines should not receive a bioresorbable scaffold.
PATIENTS PRESENTING WITH STEMI
STEMI patients are especially prone to thrombosis. Because of the increased strut thickness, which may further activate platelets, and lack of data, STEMI patients should not receive bioresorbable scaffolds.
LESIONS WITH HEAVY CALCIFICATIONS, DIFFUSE DISEASE, WITH CHALLENGING TORTUOSITY AND SEVERE ANGULATION SHOULD BE EXCLUDED
Table 4 summarises these characteristics and the recommendations reached by the expert panel. For a detailed list of contraindications, please refer to the Magmaris “instructions for use”.
PRIORITIES FOR FUTURE INVESTIGATIONS
The panel identified subsets of patients and lesions that should be investigated further:
DIABETES
Diabetic patients experience a progression of their cardiovascular disease and could be considered as an interesting subset for the use of bioresorbable scaffolds. However, diabetic patients often report smaller vessels as well as a more diffuse disease, and are more prone to inflammation.
CTO
At this stage this subset is not recommended because the sizing of the vessel is challenging, CTO lesions often being long and calcified. Current techniques to treat the lesions are complex and numerous.
BIFURCATIONS
Currently this subset is not recommended because of the overexpansion and often large overlapping needed to achieve optimal deployment with a two-stent technique. However, in case of a one-device technique, Magmaris could be considered as an option in the case of a bifurcation lesion, if evaluation by intracoronary imaging is provided.
ACUTE CORONARY SYNDROME
If infarct markers have normalised and thrombus is resolved, Magmaris could be considered as a potential device. Unresolved thrombus at the lesion site should not be treated with a magnesium bioresorbable scaffold.
FUTURE PIVOTAL STUDIES
The panel acknowledged that robust data are required to broaden the use of Magmaris. Such studies should be well powered and require an international effort. Meanwhile, post-marketing studies are planned to start after approval. A non-inferiority study of Magmaris versus Orsiro for one-year target lesion failure as the primary endpoint should be considered. While there are major differences between the Absorb design and polymer and the Magmaris, designing head-to-head trials comparing the scaffolds for superiority is premature at this stage.
Conclusion
As the first metallic-based resorbable scaffold on the market, Magmaris might potentially address some of the current shortcomings of polymer-based scaffolds. Cautious use with careful selection of patients and lesions is currently recommended to optimise the patient outcomes of the technology. This consensus document recognises that currently available data with this technology are sparse and therefore recommendations for use are restrictive at this early stage. With accumulating clinical data, recommendations for use will be more specific and broader use may be supported by the expert panel.
Conflict of interest statement
M. Haude has received study grants and lecture fees from Biotronik, Abbott Vascular, Cardiac Dimensions, Medtronic, Volcano and Lilly. M. Joner is a consultant for Biotronik, OrbusNeich, AUM Medical and reports speaker’s fees from Biotronik, OrbusNeich, Boston Scientific, Abbott Vascular and Medtronic. J. Koolen is a consultant for Biotronik. R. Tölg has received honorarium from Biotronik as well as speaker’s fees from Biotronik and Abbott Vascular. R. Waksman has received consulting fees from Boston Scientific, AstraZeneca, Abbott Vascular, Biotronik and Biosensors. He has also received fees for non-CME services from AstraZeneca, Boston Scientific and Medtronic. J. Fajadet has no conflicts of interest to declare.
