
Introduction
Magmaris® (Biotronik AG, Bülach, Switzerland) is a magnesium-based scaffold that has been successfully tested in 184 patients enrolled in the BIOSOLVE-II and BIOSOLVE-III studies [1,2. After gaining CE certification in June 2016, it was paramount to test this device in clinical routine to ensure a safe roll-out of this technology.
Methods
BIOSOLVE-IV is an international, single-arm, multicentre registry conducted in 86 centres. The first patient was enrolled in September 2016. Inclusion and exclusion criteria and endpoints are listed on ClinicalTrials.gov (NCT02817802).
Clinical follow-up was scheduled at six and 12 months, and annually up to five years (telephone follow-ups were permitted). The study was performed according to the Declaration of Helsinki and ISO14155:2011, was approved by the ethics committees, and all patients provided written informed consent. Monitoring encompassed a minimum of 25% random subjects per centre; an angiographic core laboratory assessed endpoint-related events and device failures. All potential device-related events were adjudicated by an independent clinical events committee member. In case of disagreement with a study site, the case was reviewed by a second clinical events committee member.
Magmaris has been described previously1,2. Predilatation using a non-compliant balloon with a 1:1 balloon-to-artery ratio was mandatory. The balloon should expand fully and the residual stenosis before Magmaris implantation ought to be <20%. The scaffold implantation ought to follow the recommendation of the instructions for use and the consensus letter of Fajadet et al3. Post-dilatation with a non-compliant balloon at high pressure (>16 atm) was recommended. Dual antiplatelet therapy (DAPT) was recommended for at least six months.
The sample size of 2,054 patients was calculated based on the null hypothesis that Magmaris has a 12-month probable or definite scaffold thrombosis rate of ≥1.49%. Endpoint analyses were planned after every 200 subjects reaching the primary endpoint.
Results
This publication refers to the first 400 patients with 425 lesions enrolled. Patient age ranged from 29 to 86 years (Table 1); 63 patients (15.8%) presented with non-ST-elevation myocardial infarction (NSTEMI) (Figure 1). Predilatation and post-dilatation were performed in nearly all patients. Magmaris could not be implanted in three patients (0.8%) (Table 2).
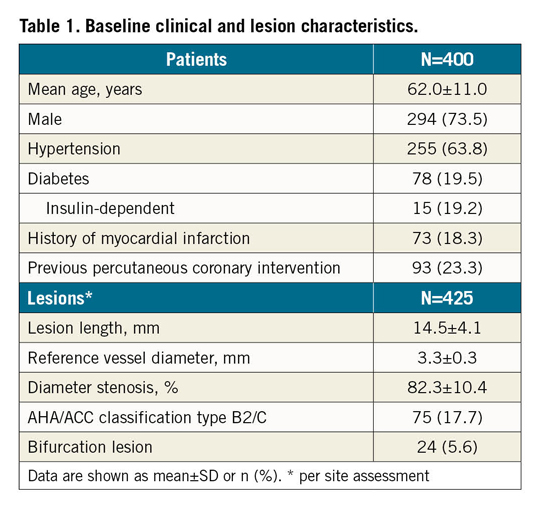
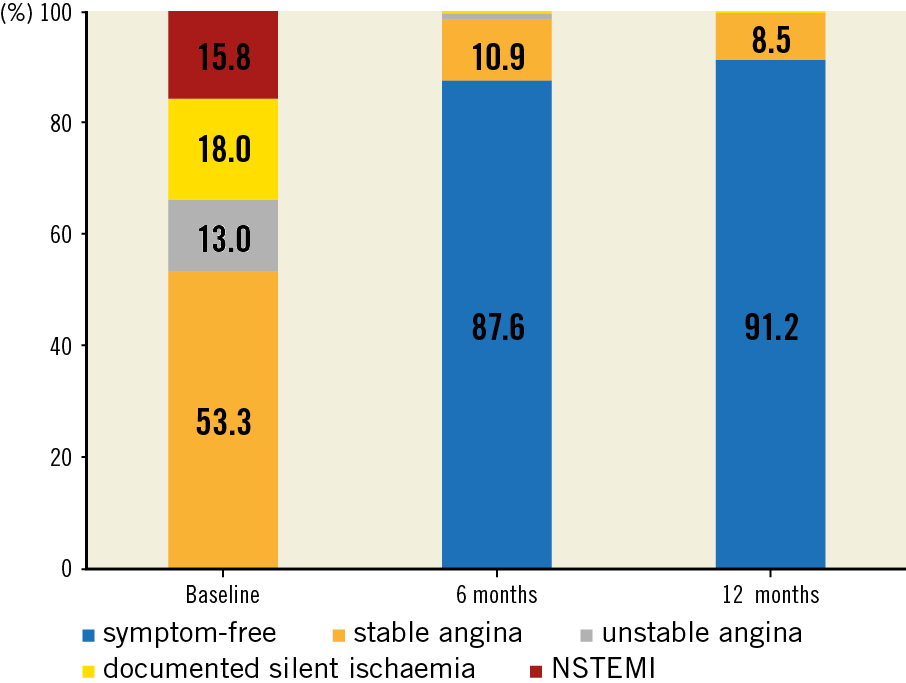
Figure 1. Ischaemic status at baseline and follow-up. NSTEMI: non-ST-elevation myocardial infarction
Concerning 12-month follow-up data, two patients had died of cancer, six visits were missed and one patient withdrew consent (compliance of 98.2%, 390/397); 76.8% (298/388) were on DAPT. The primary endpoint, target lesion failure (TLF) at 12 months, was 4.3% (n=17) (Figure 2). All had clinically driven target lesion revascularisation (TLR), of whom three (0.8%) had additional target vessel myocardial infarction (TV-MI).
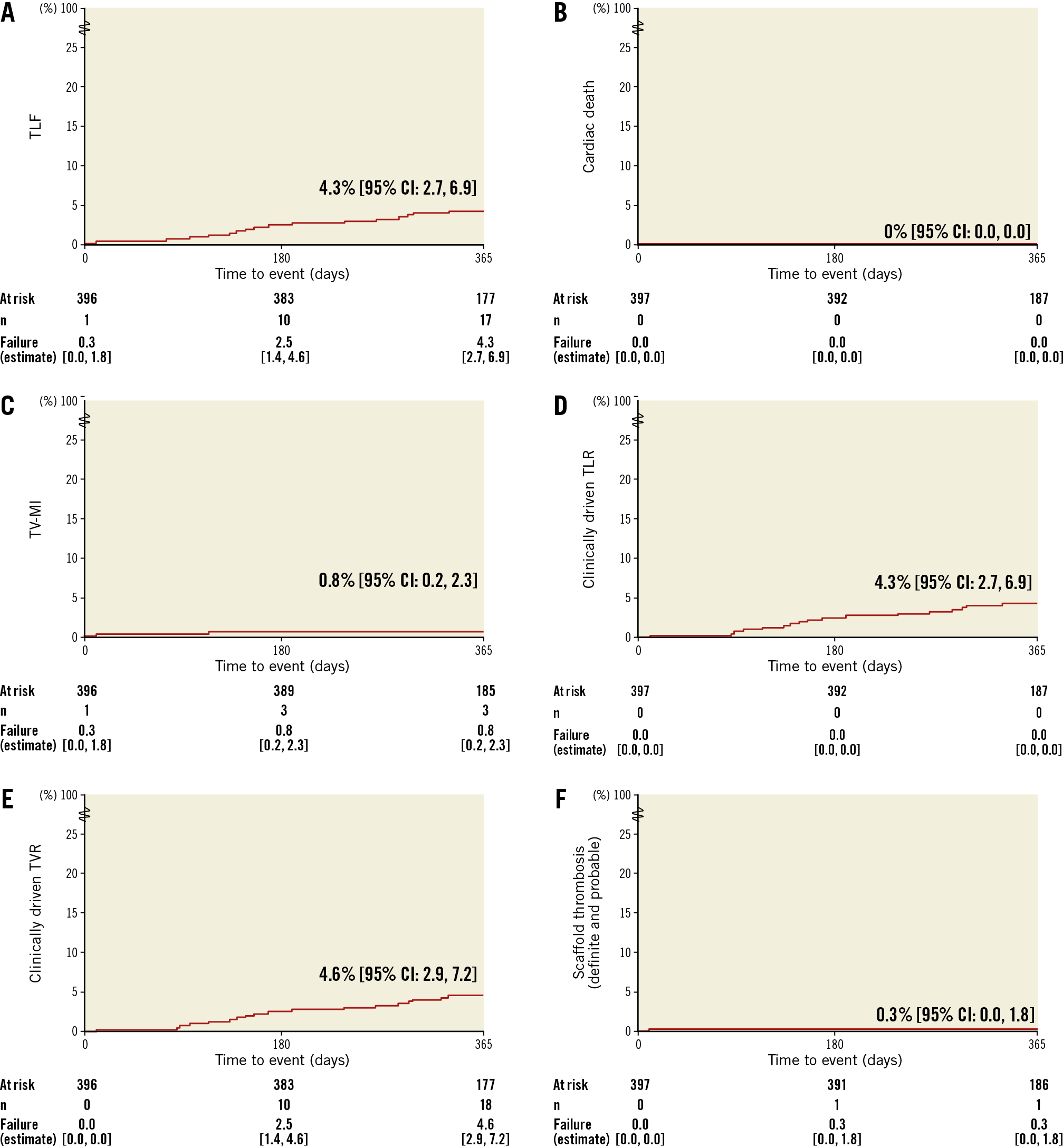
Figure 2. Kaplan-Meier estimates of clinical outcomes up to 12 months. A) TLF. B) Cardiac death. C) TV-MI. D) Clinically driven TLR. E) Clinically driven TVR. F) Definite and probable scaffold thrombosis. TLF: target lesion failure; TLR: target lesion revascularisation; TV-MI: target vessel myocardial infarction (periprocedural MI were adjudicated using SCAI definitions and spontaneous MI using the extended historical definitions); TVR: target vessel revascularisation
One definite scaffold thrombosis (0.3%) occurred on post-procedure day 10. The patient was admitted with NSTEMI and severe three-vessel coronary artery disease. First, the occluded circumflex artery was treated using a non-study stent. Four days later, the index procedure with the Magmaris scaffold was performed to treat a stenosis in a heavily calcified right coronary artery. Thereafter, a minimally invasive direct coronary artery bypass graft (left internal mammary artery to ramus interventricularis anterior) was intended and hence DAPT was interrupted five days after the index procedure. Five days later, a scaffold thrombosis occurred that led to a TV-MI and required a TLR. The patient was alive at 12 months.
Discussion
This pre-specified interim analysis of the first 400 patients enrolled in the BIOSOLVE-IV registry confirmed the favourable safety outcomes of the BIOSOLVE-II and BIOSOLVE-III trials with low TLF rates at 12 months comparable to new-generation permanent drug-eluting stents (DES), no cardiac death and only one scaffold thrombosis that occurred after DAPT interruption.
Comparing outcomes to the BIOSOLVE-II and BIOSOLVE-III studies, in BIOSOLVE-IV, baseline characteristics were similar with relatively simple lesions. The only relevant difference was the inclusion of 15.8% NSTEMI patients who were excluded in the precursor studies1,2. The rate of TLF was also similar, albeit clinically driven TLR was slightly higher in BIOSOLVE-IV. One definite scaffold thrombosis occurred in our series, which is the first definite scaffold thrombosis in the 584 patients published to date1,2. It occurred at day 10 after DAPT cessation in a patient with severe three-vessel disease after a staged procedure, emphasising that DAPT in the early days after Magmaris implantation is as important as after DES implantation.
Our outcomes are also within the range of the objective performance criteria for new-generation stents as postulated by the ESC/EAPCI’s task force, namely 2.91% TLR (IQR 1.67-5.94) and 0.47% definite stent thrombosis (IQR 0.28–0.72) at 9- to 12-month follow-up4.
Limitations
BIOSOLVE-IV has limitations inherent to observational registries.
Conclusion
This preliminary analysis after 400 patients enrolled in the BIOSOLVE-IV real-world registry confirms the safe roll-out of a magnesium-based bioresorbable scaffold into clinical practice with low TLF rates and only one scaffold thrombosis after DAPT cessation.
|
Impact on daily practice This registry represents the largest clinical experience with the sirolimus-eluting magnesium-based Magmaris BRS to date. It provides additional assurance on the safety and performance of the device with low TLF and stent thrombosis rates. DAPT cessation prior to six months should be strongly discouraged. |
Acknowledgements
We thank MedStar Health Research Institute for angiographic core laboratory services and Beatrix Doerr for her help in drafting this manuscript, reimbursed by Biotronik AG.
Funding
This study was funded by Biotronik AG, Bülach, Switzerland.
Conflict of interest statement
S. Verheye reports receiving speaking fees from Alvimedica, Biotronik, and Elixir Medical. M. Haude reports receiving study grants and personal fees from Biotronik, Abbott Vascular, Cardiac Dimensions, and Philips. A. Aminian reports receiving grants from Biotronik. J. Bennett has received research grants and speaking fees from Abbott Vascular and Biotronik AG. The other authors have no conflicts of interest to declare.
