Abstract
Background: The third-generation coronary sirolimus-eluting magnesium scaffold, DREAMS 3G, is a further development of the DREAMS 2G (commercial name Magmaris), aiming to provide performance outcomes similar to drug-eluting stents (DES).
Aims: The BIOMAG-I study aims to assess the safety and performance of this new-generation scaffold.
Methods: This is a prospective, multicentre, first-in-human study with clinical and imaging follow-up scheduled at 6 and 12 months. The clinical follow-up will continue for 5 years.
Results: A total of 116 patients with 117 lesions were enrolled. At 12 months, after completion of resorption, in-scaffold late lumen loss was 0.24±0.36 mm (median 0.19, interquartile range 0.06-0.36). The minimum lumen area was 4.95±2.24 mm² by intravascular ultrasound and 4.68±2.32 mm² by optical coherence tomography. Three target lesion failures were reported (2.6%, 95% confidence interval: 0.9-7.9), all clinically driven target lesion revascularisations. Cardiac death, target vessel myocardial infarction and definite or probable scaffold thrombosis were absent.
Conclusions: Data at the end of the resorption period of DREAMS 3G showed that the third-generation bioresorbable magnesium scaffold is clinically safe and effective, making it a possible alternative to DES. ClinicalTrials.gov: NCT04157153.
Introduction
Resorbable scaffolds were developed to avoid the long-term adverse outcomes associated with the implantation of permanent metallic drug-eluting stents (DES). They disappear after the initial healing phase of the vessel, thus preventing long-term straightening, which may have a positive effect on wall shear stress1. Magnesium is an attractive bioresorbable material because of its mechanical properties, which are similar to those of conventional DES123.
The second-generation CE (European conformity)-marked Drug-Eluting Resorbable Magnesium Scaffold (DREAMS 2G, commercial name Magmaris; BIOTRONIK) showed very good outcomes in multiple trials, but angiographic in-scaffold late lumen loss (LLL) was higher than observed with contemporary DES245. Serial imaging analyses of DREAMS 2G have shown that LLL was not only associated with neointimal hyperplasia, but also with constrictive remodelling. Therefore, a new-generation sirolimus-eluting resorbable magnesium coronary scaffold (DREAMS 3G) was developed, which has an improved scaffold material providing a substantially increased radial force, thinner struts and prolonged scaffolding time while maintaining the resorption time of 1 year36.
The prospective, international, multicentre, first-in-human clinical trial, BIOMAG-I, now aims to assess the angiographic and intracoronary imaging results as well as the safety and clinical performance of DREAMS 3G in humans. Six-month data have been reported previously6; we herein report the 12-month clinical and imaging data, which represent outcomes after the complete resorption of DREAMS 3G.
Methods
STUDY DESIGN AND PATIENTS
The study methods have been reported in detail previously, and the clinical study protocol is available as supplementary material in the publication of the 6-month results6. In brief, the prospective, multicentre, single-arm, first-in-human study was conducted in 8 countries in Europe. The main inclusion criteria were symptomatic coronary artery disease, a maximum of 2 de novo single lesions in 2 separate coronary arteries, and reference vessel diameters ranging from 2.5 mm to 4.2 mm with a maximum lesion length of ≤28 mm. The main exclusion criteria were ST-elevation myocardial infarction, unsuccessful predilatation, left main stenosis, or chronic total occlusion. The full list of inclusion and exclusion criteria is available at ClinicalTrials.gov: NCT04157153.
The study was conducted according to the current version of the Declaration of Helsinki, ISO14155, and local guidelines and regulations, and was approved by the ethics committee of each centre. All patients provided written informed consent before any study procedure. Thorough study oversight was ensured through monitoring with 100% source document verification, involvement of a steering committee, an independent clinical events committee that adjudicated all endpoint-related events, and an independent core laboratory (for angiographic assessment, intravascular ultrasound [IVUS], and optical coherence tomography [OCT]).
STUDY PROCEDURES
The DREAMS 3G system consists of a balloon-expandable scaffold mounted on a rapid-exchange delivery system. The scaffold is made from a proprietary magnesium alloy (BIOmag-alloy) that includes aluminium and magnesium. The strut thicknesses are 99 μm for device diameter 2.5 mm, 117 μm for device diameters 3.0 mm and 3.5 mm, and 147 μm for device diameter 4.0 mm. Subsequently, the total surface area is slightly reduced compared to its precursor, DREAMS 2G (from 9.2 mm² to 7.8 mm² per mm scaffold length). The resorption of the scaffold is completed within 12 months36. The scaffold backbone is coated with poly-L-lactic acid (PLLA) incorporating sirolimus at a concentration of 1.4±0.3 μg per mm26.
Implantations had to follow the criteria of the instructions for use and the consensus from the expert panel published by Fajadet et al7. These include adequate patient and lesion selection (e.g., excluding patients in whom a full expansion of the predilatation balloon cannot be achieved, patients with thrombus at the lesion site, patients for whom a return of vasomotion cannot be expected, patients for whom proper sizing cannot be achieved, left main lesions, dual antiplatelet therapy contraindications, ST-elevation myocardial infarction, lesions with heavy calcification, diffuse disease, and challenging tortuosity and severe angulation, thus excluding lesions with a high risk of acute or late recoil), proper sizing (lesion size and length should be carefully assessed to match the matrix of device sizes and lengths), adequate predilatation (non-compliant balloon, 1:1 balloon-to-artery ratio, residual stenosis prior to implantation ≤20%), and adequate post-dilatation (non-compliant balloon ≤0.5 mm larger than the implanted nominal scaffold and expanded at >16 atm). A second DREAMS 3G was permitted in case of incomplete lesion coverage or dissection but had to be placed end-to-end7 and not overlapping. Dual antiplatelet therapy was recommended for at least 6 months.
Clinical follow-up was scheduled at 1, 6, 12, 24, 36, 48 and 60 months, additionally, imaging follow-up was performed at 6 and 12 months. These included angiographic, OCT, and IVUS assessments. Details of the image acquisition and assessments have been provided previously6.
OUTCOMES
The primary endpoint, in-scaffold LLL at 6 months, was reported previously6. Secondary endpoints at 12 months were angiographic in-scaffold and in-segment LLL, binary restenosis and diameter stenosis, and a descriptive analysis of IVUS and OCT parameters. Clinical endpoints were target lesion failure (TLF) and its subcomponents (cardiac death, target vessel myocardial infarction89, and clinically driven target lesion revascularisation), clinically driven target vessel revascularisation, and definite and probable scaffold thrombosis10.
STATISTICAL ANALYSIS
The sample size was calculated based on the primary endpoint, in-scaffold LLL at 6 months, and has been reported previously6. A second calculation was performed for the secondary endpoint, in-scaffold LLL at 12 months, comparing DREAMS 3G with data from its precursor, DREAMS 2G, and other bioresorbable scaffolds (Supplementary Table 1)11121314, resulting in a weighted mean of 0.33 mm for in-scaffold late lumen loss and a weighted pooled standard deviation (SD) of 0.35 mm. Considering a prespecified non-inferiority margin of 0.145 mm, a power of 95%, an alpha of 0.025, and a dropout rate of 25%, it was calculated that 104 patients needed to be enrolled in the study.
Outcomes are based on the intention-to-treat population and the available data. Normal distribution was assessed with the Shapiro-Wilk test. Continuous variables are expressed as means with SD and medians with interquartile ranges (IQR), as applicable. Categorical variables are expressed as absolute and relative frequencies. Kaplan-Meier estimates were used for time-to-event analysis and are presented with 95% confidence intervals (CI). Comparisons between baseline and follow-up were performed in paired data using the t-test. The statistical analysis was performed using SAS software version 9.4 (SAS Institute).
Results
BIOMAG-I enrolled 116 patients between April 2020 and February 2022 (Figure 1).
Baseline and procedural data have been published previously6. In brief, patients were 61.0±9.0 years on average, 77.8% were male, 74.1% had hypertension, 62.1% had hypercholesterolaemia, 64.7% had a history of smoking, 27.6% had diabetes, 33.6% had a previous myocardial infarction, and 20.7% presented with non-ST-elevation myocardial infarction (NSTEMI). Lesions (N=117) were 12.3±5.1 mm long with a reference vessel diameter of 2.72±0.46 mm, 76.9% were Type B2/C, and 2.6% of lesions were moderate or severely calcified.
Pre- and post-dilatation were performed in all lesions. Device success − defined as a final residual diameter stenosis of <30% by quantitative coronary angiography (QCA) or visual assessment using the assigned device, successful delivery of the scaffold to the target lesion, appropriate scaffold deployment, and successful removal of the delivery system − was obtained in 97.7% (126/129) of devices (Supplementary Table 2). Procedural success − defined as a final diameter stenosis of <30% by QCA, using any percutaneous method, without the occurrence of death, Q-wave or non-Q-wave myocardial infarction, or TLR during the hospital stay − was achieved in 99.1% (115/116) of patients.
Serial QCA data could be obtained in 100 patients. Paired in-scaffold LLL was 0.19±0.25 mm (95% CI: 0.14-0.24, median 0.13 [IQR: 0.04-0.32]) at 6 months and 0.24±0.36 mm (95% CI: 0.17-0.31, median 0.19 [IQR: 0.06-0.36] at 12 months (Table 1, Central illustration). Thus, the null hypothesis was rejected, and non-inferiority to the historical control of resorbable scaffolds was demonstrated.
Serial IVUS and OCT data were available in 75 and 89 patients, respectively (for unpaired data, see Supplementary Table 3). IVUS assessment showed the minimum lumen and device areas were 4.98±1.99 mm² at 6 months versus 4.95±2.24 mm² at 12 months and 5.01±1.97 mm² at 6 months versus 5.01±2.29 mm² at 12 months, respectively, and the mean plaque area regressed from 7.90±2.77 mm² at 6 months to 7.46±2.65 mm² at 12 months, p=0.0003 (Table 1, Figure 2).
By OCT, no intraluminal mass was observed at any time, and at 12 months, the struts were no longer discernible (Table 1, Central illustration).
At 12 months, 76.3% (87/114) of patients were still on dual antiplatelet therapy (Supplementary Table 4), no patient had an acute coronary syndrome, 17.5% (20/116) of patients had stable angina, and 3.5% (4/116) had documented silent ischaemia.
Clinical follow-up was available for 98.3% (114/116) of the patients. The Kaplan-Meier estimate for 12-month TLF was 2.6% (95% CI: 0.9-7.9) (Figure 3), consisting of 3 clinically driven TLRs. The first occurred on day 166 after implantation (using a 3.5x30 mm device) in an asymptomatic patient with 63% diameter stenosis and an instantaneous wave-free ratio of 0.51. The original lesion was classified as very fibrotic by the core laboratory, and analysis showed that the pre- and post-dilatation was not sufficient for this type of lesion. The second TLR occurred on day 204 in a patient that was treated with two 2.5x13 mm devices because of a dissection that occurred during the implantation of the first device. By core laboratory assessment, the reference vessel diameter (RVD) was 1.88 mm. The patient presented with atypical chest pain and a 51% diameter stenosis. The third TLR occurred on day 270 in a lesion with a high plaque burden treated with a 4.0x22 mm device. The patient presented as asymptomatic at 6 months, but with an in-device LLL of 1.83 mm. An angiographic control on day 270 revealed a 77% diameter stenosis with a LLL of 2.65 mm that was treated with a DES (Supplementary Table 2).
One additional clinically driven target vessel revascularisation occurred during the scheduled 6-month angiography because of ostial target vessel dissection during guide catheter positioning. No cardiac death, myocardial infarction or probable scaffold thrombosis were reported.
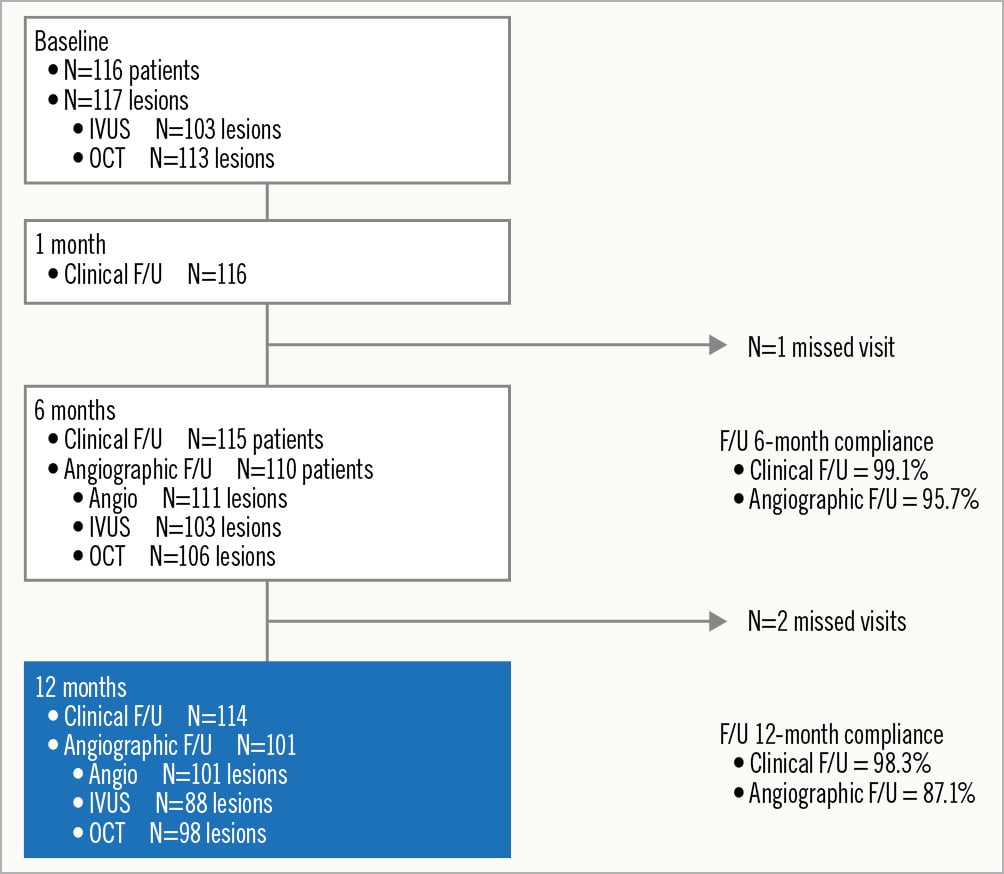
Figure 1. Patient flowchart. A total of 116 patients with 117 lesions were enrolled. At 12 months, serial data (reflecting preprocedure, post-procedure and 6 and 12 months) were available for 100 lesions with angiographic follow-up, 75 lesions with intravascular ultrasound (IVUS) follow-up and for 89 lesions with optical coherence tomography (OCT) follow-up. F/U: follow-up
Table 1. Core laboratory assessed imaging analysis – paired data.
| Preprocedure | Post-procedure | 6 M | 12 M | ∆ post-procedure vs 12 M | p-value post-procedure vs 12 M | ∆ 6 M vs 12 M | p-value 6 M vs 12 M | |
|---|---|---|---|---|---|---|---|---|
| Angiography | N=100 | N=100 | N=100 | N=100 | N=100 | N=100 | N=100 | N=100 |
| RVD, mm | ||||||||
| In-scaffold | NA | 2.86±0.462.83 (2.54-3.21) | 2.86±0.492.81 (2.49-3.18) | 2.83±0.522.80 (2.39-3.17) | −0.03±0.33 | 0.384 | −0.03±0.24 | 0.250 |
| In-segment | 2.74±0.462.71 (2.42-3.06) | 2.77±0.502.74 (2.42-3.08) | 2.81±0.512.79 (2.46-3.14) | 2.74±0.522.63 (2.35-3.15) | −0.03±0.36 | 0.356 | −0.07±0.29 | 0.015 |
| MLD, mm | ||||||||
| In-scaffold | NA | 2.61±0.432.59 (2.27-2.91) | 2.41±0.492.40 (2.06-2.72) | 2.37±0.532.34 (2.01-2.67) | −0.24±0.36 | <0.0001 | −0.05±0.19 | 0.014 |
| In-segment | 1.09±0.391.03 (0.84-1.32) | 2.33±0.452.35 (2.03-2.56) | 2.29±0.46 2.21 (1.94-2.56) | 2.23±0.502.17 (1.90-2.52) | -0.10±0.42 | 0.015 | -0.06±0.27 | 0.024 |
| DS, % | ||||||||
| In-scaffold | NA | 8.6±5.58.0 (5.0-11.0) | 15.4±9.814.0 (8.0-20.0) | 16.31±10.5314.0 (10.0-21.0) | 7.7±11.0 | <0.0001 | 0.94±7.95 | 0.243 |
| In-segment | 60.4±12.661.0 (51.5-69.0) | 15.5±8.413.5 (10.0-21.0) | 18.2±9.617.0 (11.3-23.0) | 18.5±10.917.0(12.0-23.0) | 2.94±13.97 | 0.038 | 0.27±9.62 | 0.784 |
| Binary restenosis, % | ||||||||
| In-scaffold | NA | NA | 0 (0%) | 1 (1.0%) | NA | NA | NA | NA |
| In-segment | NA | NA | 0 (0%) | 1 (1.0%) | NA | NA | NA | NA |
| LLL, mm | ||||||||
| In-scaffold | NA | NA | 0.19±0.250.13 (0.04-0.32) | 0.24±0.360.19 (0.06-0.36) | NA | NA | −0.05±0.19 | 0.014 |
| In-segment | NA | NA | 0.04±0.330.06 (-0.17 to 0.22) | 0.10±0.420.12 (-0.08 to 0.25) | NA | NA | −0.06±0.27 | 0.024 |
| IVUS | N=75 | N=75 | N=75 | N=75 | N=75 | N=75 | N=75 | N=75 |
| Mean vessel area, mm² | 12.88±4.5211.67 (9.19-15.63) | 15.19±4.5614.22 (12.00-17.81) | 14.93±4.7714.20 (11.41-16.77) | 14.59±4.9113.52 (11.23-16.39) | −0.60±1.94 | 0.009 | −0.34±1.41 | 0.039 |
| Mean scaffold area, mm² | NA | 7.63±2.187.20 (6.15-8.90) | 7.04±2.416.83 (5.32-8.14) | 7.13±2.74 6.50 (5.13-8.58) | −0.50±1.47 | 0.004 | 0.09±0.86 | 0.354 |
| Minimum scaffold area, mm² | NA | 6.47±2.016.13 (5.25-7.89) | 5.01±1.974.76 (3.58-5.74) | 5.01±2.294.28 (3.37-6.09) | −1.45±1.44 | <0.0001 | 0.01±1.01 | 0.962 |
| Mean lumen area, mm² | 5.96±2.255.50 (4.23-7.40) | 7.65±2.237.20 (6.15-9.00) | 7.00±2.426.73 (5.31-8.09) | 7.09±2.736.48 (5.11-8.59) | −0.56±1.45 | 0.001 | 0.05±0.85 | 0.593 |
| Minimum lumen area, mm² | 3.23±1.332.83 (2.39-3.71) | 6.47±2.026.13 (5.25-7.89) | 4.98±1.994.72 (3.51-5.74) | 4.95±2.244.24 (3.37-6.09) | −1.52±1.43 | <0.0001 | −0.03±1.04 | 0.807 |
| Mean plaque area, mm² | 6.89±3.266.22 (4.72-8.50) | 7.48±2.856.72 (5.63-9.20) | 7.90±2.777.24 (6.05-9.78) | 7.46±2.656.93 (5.57-8.97) | −0.02±1.22 | 0.911 | −0.44±1.0 | 0.0003 |
| Mean NIH area, mm² | NA | NA | 0.09±0.21 | 0.14±0.24 | NA | NA | 0.06±0.09 | |
| Total incomplete strut apposition area*, mm² | NA | 0.15±0.220.07 (0.02-0.21) | 0.07±0.080.06 (0.02-0.07) | 0.03±0.040.02 (0.00-0.05) | –0.11±0.13 | 0.136 | –0.05±0.07 | 0.050 |
| Number of struts | NA | 211.6±74.1209.5 (167.0-259.0) | NA† | NA† | NA† | NA | NA† | NA |
| OCT | N=89 | N=89 | N=89 | N=89 | N=89 | N=89 | N=89 | N=89 |
| Mean scaffold area, mm² | NA | 8.80±2.488.28 (6.84-10.6) | NA† | NA† | NA† | NA | NA† | NA |
| Mean lumen area, mm² | 5.68±2.045.51 (4.14-6.98) | 8.75±2.488.26 (6.8-10.5) | 7.02±2.536.31 (5.21-8.27) | 7.03±2.766.75 (4.89-8.43) | –1.73±1.84 | <0.0001 | 0.01±1.10 | 0.955 |
| MLD, mm | 1.32±0.341.29 (1.04-1.58) | 2.66±0.432.63 (2.32-3.02) | 2.12±0.492.05 (1.83-2.40) | 2.09±0.522.03 (1.72-2.36) | 0.57±0.47 | <0.0001 | –0.03±0.29 | 0.289 |
| Minimum lumen area, mm² | 2.08±0.901.99 (1.37-2.59) | 7.28±2.216.99 (5.47-9.0) | 4.85±2.214.23 (3.31-5.94) | 4.68±2.323.92 (3.12-5.83) | −2.60±1.81 | <0.0001 | −0.17±0.96 | 0.094 |
| Malapposed struts,% | NA | 4.62±4.693.68 (0.79-6.80) | NA† | NA† | NA† | NA† | NA† | NA† |
| Total incomplete strut apposition area, mm² | NA | 0.08±0.110.05 (0.01-0.12) | NA† | NA† | NA† | NA† | NA† | NA† |
| Total tissue protrusion, mm2 | NA | 0.20±0.130.19 (0.12-0.26) | NA† | NA† | NA† | NA† | NA† | NA† |
| Data are mean±SD, median (IQR), or n (%). *Non-serial data (malapposition area is only measured if it is present). †Struts were barely discernible at 6 months and no longer discernible at 12 months by OCT; only strut remnants were observed by IVUS at 12 months. DS: diameter stenosis; IQR: interquartile range; IVUS: intravascular ultrasound; LLL: late lumen loss; M: months; MLD: minimum lumen diameter; NA: not applicable; NIH: neointimal hyperplasia; OCT: optical coherence tomography; RVD: reference vessel diameter; SD: standard deviation | ||||||||
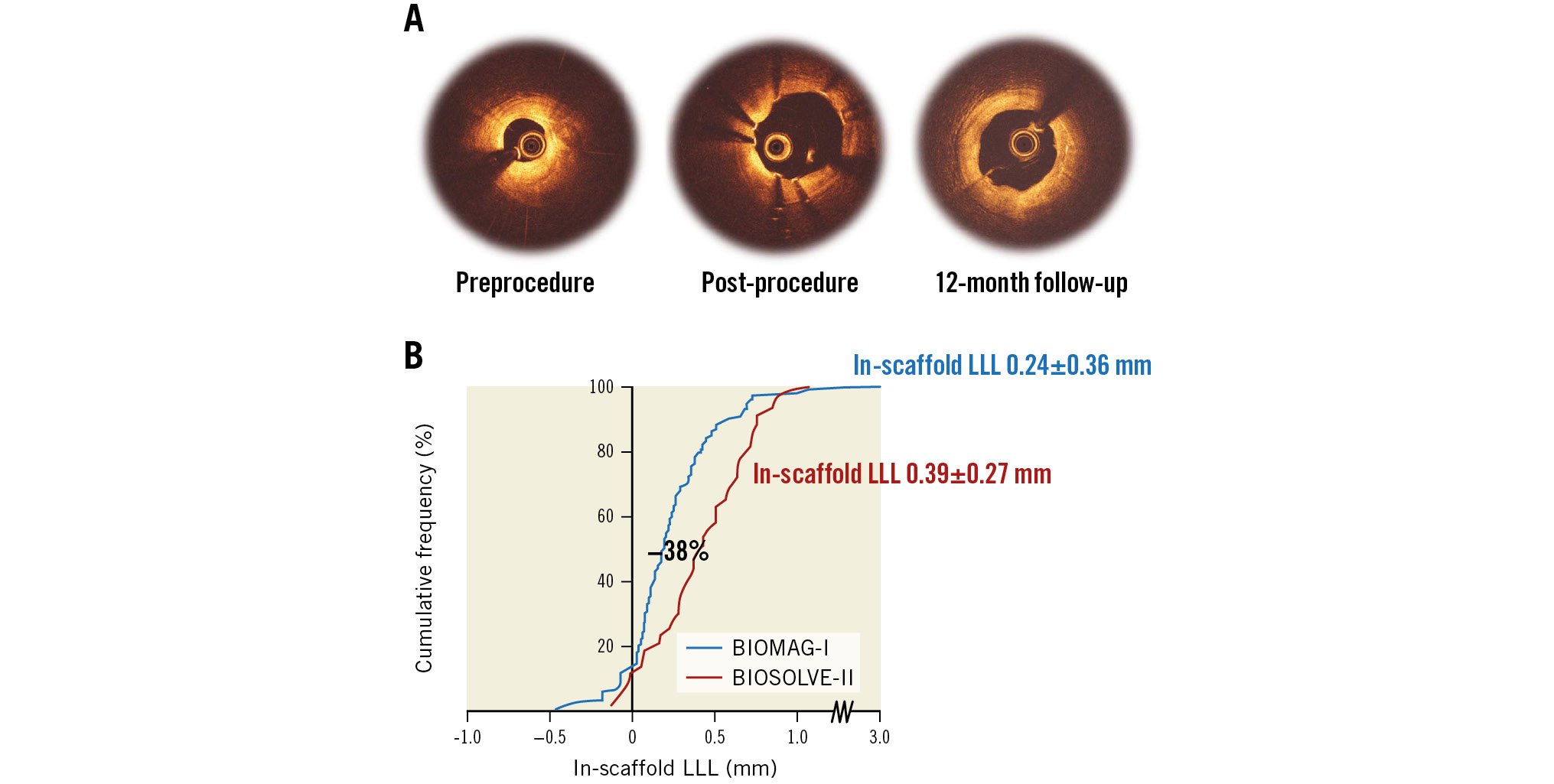
Central illustration. Optical coherence tomography of strut apposition and absorption, and in-device late lumen loss measured by quantitative coronary angiography. A) Optical coherence tomography showed good postprocedural strut apposition and that the struts were no longer discernible at 12 months. B) The in-scaffold late lumen loss (LLL) improved by 38% compared to the precursor of DREAMS 3G, the DREAMS 2G, in the BIOSOLVE-II study.
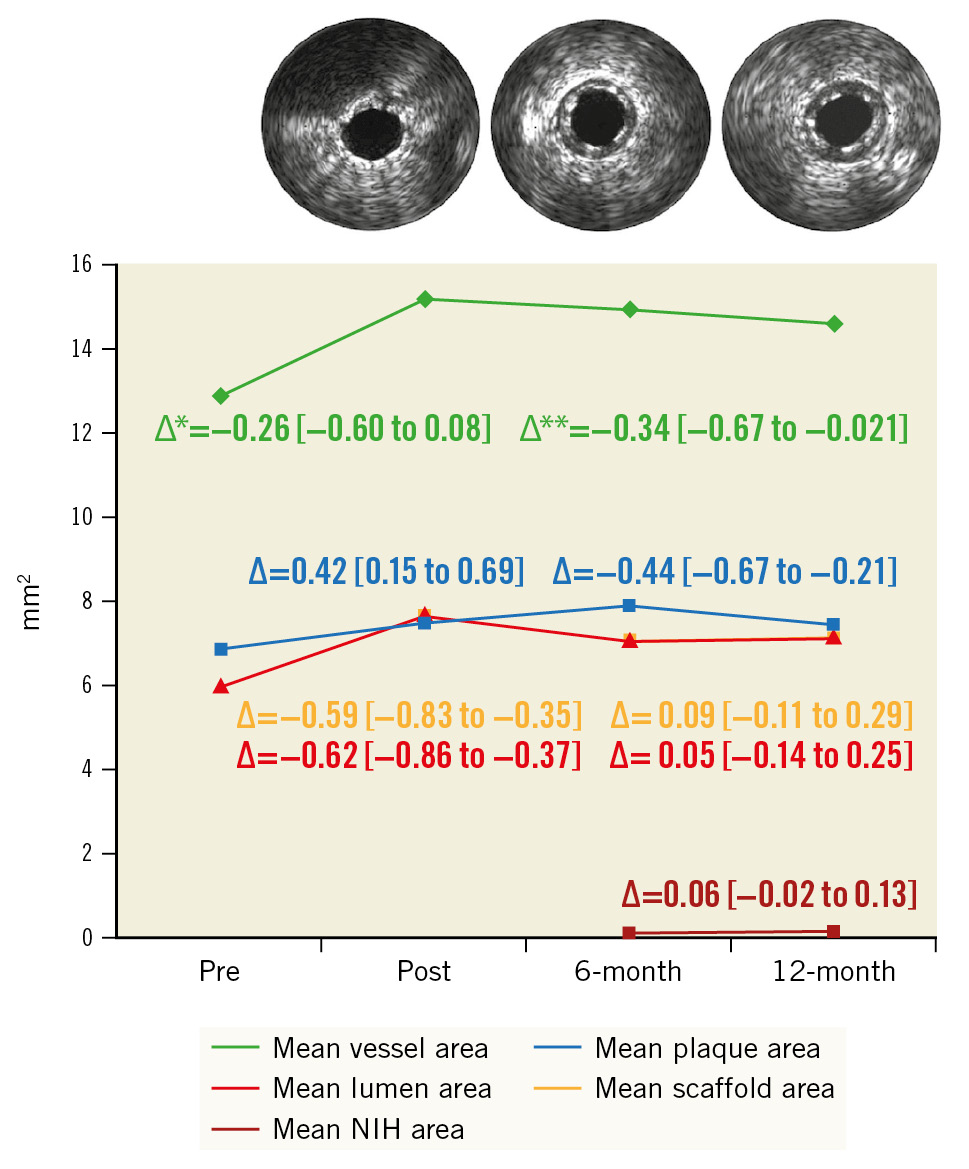
Figure 2. Serial area changes by intravascular ultrasound. Paired intravascular ultrasound data were available for 75 patients (core laboratory analysed). The mean lumen area and the mean scaffold area are nearly identical. Subsequently the different curves are not discernible. ∆ indicates the difference between follow-ups in mm2 [95% CI]. Δ* refers to post-procedure versus six months, and Δ** refers to 6 months versus 12 months. CI: confidence interval; NIH: neointimal hyperplasia
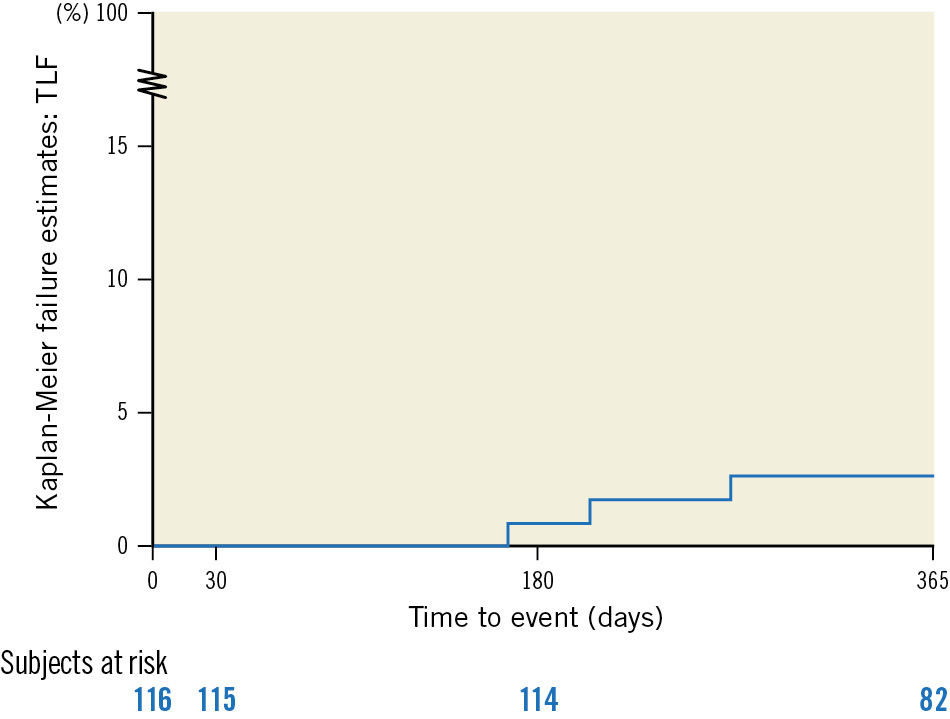
Figure 3. Target lesion failure at 12 months per Kaplan-Meier analysis. All 3 target lesion failures were clinically driven target lesion revascularisations; no target vessel myocardial infarction nor cardiac death was reported. TLF: target lesion failure
Discussion
These are the first 12-month data presented for the new-generation DREAMS 3G scaffold, which represent the outcomes at the time point of complete resorption3.
While there is a large body of evidence confirming that implantation of the precursor of DREAMS 3G, DREAMS 2G, resulted in low (long-term) clinical event rates, the angiographic parameters such as in-scaffold LLL were not competitive with contemporary DES245141516. The DREAMS 3G was developed to maintain the overall resorption time, to improve the radial strength and to prolong the scaffolding time to prevent constrictive remodelling and to achieve LLL values similar to those of contemporary DES. Therefore, qualitative and quantitative aspects of the degradation were enhanced using a new magnesium alloy, resulting in more homogeneous resorption and prolonged stability. The new magnesium alloy even permitted a reduction in strut thickness down to 99 μm for the smallest device diameter and, subsequently, a reduction in the total surface area compared to DREAMS 2G. This is of relevance, as strut thickness is associated with restenosis1718.
These new features were tested in a porcine animal model, where the discontinuity density of DREAMS 3G over time was smaller than for DREAMS 2G, reflecting an improved radial strength. Furthermore, DREAMS 3G exhibited a more homogeneous strut degeneration with less variability3.
The serial data presented herein confirm the design goals. While there was a significant increase in in-device LLL between 6 and 12 months (from 0.19±0.25 mm to 0.24±0.36 mm; p=0.014), the change is not seen as clinically relevant, as it was below the spatial resolution of angiography19. Yet, it could be interpreted as a sign that the scaffold resorption is not completed at 6 months and that a certain loss of radial strength may occur between 6 and 12 months, corresponding to what was observed in the preclinical testing with scaffold resorption of 64.9% at 6 months3.
Most relevant, the in-device LLL at the end of the resorption period at 12 months was 0.24±0.36 mm (median 0.19, IQR: 0.06-0.36), thus 38% lower than the 0.39±0.27 mm reported in the BIOSOLVE-II trial, with the caveat that the 4P-principles7 were not fully applied at the time of patient inclusion in BIOSOLVE-II2. Furthermore, the LLL is within the range of contemporary DES with a median of 0.18 mm (IQR 0.13-0.25) reported for new-generation DES at 9 months by the European Society of Cardiology/European Association of Percutaneous Cardiovascular Interventions (ESC/EAPCI) task force20.
As seen with DREAMS 2G in BIOSOLVE-II, intraluminal mass was absent at all time points, and at 12 months, strut-like remnants were visible by IVUS but not by OCT2.
In terms of clinical data, 3 clinically driven TLRs occurred, resulting in a 12-month rate of 2.6%. No myocardial infarction and no definite or probable scaffold thrombosis occurred during 12 months, despite the recommendation for dual antiplatelet therapy for 6 months only. Considering the full resorption of the scaffold, it is not expected that there will be any thrombotic events related to the device remnants beyond 12 months. The properties of the magnesium scaffold that are protective for scaffold thrombosis have been summarised in detail previously2 and include a negatively charged surface with antithrombotic properties, laser polishing and rounded edges, a resorption period of only 12 months, and metal-like behaviour during implantation, resulting in better expansion and apposition2122.
These good angiographic and clinical outcomes might have also been impacted by the 4P-principles that were largely adhered to, as detailed in the 6-month publication67. Although, 2 out of the 3 patients with clinically driven TLR did not adhere to the 4P-principles. With an RVD of 1.88 mm, 1 patient was in violation of the inclusion criteria of RVDs between 2.5 and 4.0 mm. The second patient had insufficient pre- and post-dilatation. This emphasises that the 4P-principles should be strictly adhered to.
Limitations
Limitations include those inherent to single-arm studies that limit the comparison to other devices. Furthermore, despite including a high percentage of Type B2/C lesions and NSTEMI patients, the population still does not reflect the overall PCI population in daily practice. A subgroup analysis by device diameter would have been interesting; however, the subgroup sample sizes were too small to provide meaningful outcomes.
Conclusions
With the caveat that very complex lesions were excluded, the initial results from the BIOMAG-I first-in-human trial showed that the third-generation drug-eluting resorbable magnesium scaffold, DREAMS 3G, met its design goals. It has an improved LLL compared to its precursor, the DREAMS 2G. Intravascular imaging revealed good strut apposition and lumen preservation between 6 and 12 months. Furthermore, struts were no longer discernible by OCT, and only strut remnants were found by IVUS, confirming scaffold resorption. The excellent safety profile of the previous generation of DREAMS was maintained in DREAMS 3G, with low TLF rates and an absence of target vessel myocardial infarction and definite or probable scaffold thrombosis, making DREAMS 3G a potential alternative to permanent DES, avoiding lifelong metallic implants associated with adverse long-term outcomes. These outcomes will need to be confirmed in large randomised clinical trials comparing DREAMS 3G with contemporary DES.
Impact on daily practice
BIOMAG-I is the first trial to report outcomes of the new-generation sirolimus-eluting bioresorbable magnesium scaffold, DREAMS 3G. It shows low angiographic in-scaffold LLL and excellent clinical safety and efficacy outcomes at 1 year after implantation, which represents the end of the device resorption period. With these results, DREAMS 3G can emerge as a competitive alternative to contemporary DES.
Acknowledgements
Data monitoring committee: Ralf Birkemeyer, Lutz Büllesfeld (St. Marien Hospital, Bonn, Germany), Bernhard Witzenbichler (Helios Amper-Klinikum Dachau, Dachau, Germany), Steffen Schneider (DSMB only, Institut für Herzinfarktforschung, Ludwigshafen, Germany). Steering Committee: Chair: Ron Waksman (MedStar Washington Hospital Center, Washington, D.C., USA), Michael Haude, (Rheinland Klinikum Neuss GmbH, Lukaskrankenhaus, Germany), Michael Joner (Deutsches Herzzentrum München, München, Germany), Stefano Galli (Centro Cardiologico Monzino, Milan, Italy). Consultant medical writer: Beatrix Doerr (Moosburg, Germany). Statistician: Christian Knoll (BIOTRONIK AG, Buelach, Switzerland). Core laboratory support: Solomon Beyene and Gebremedhin Melaku (MedStar Washington Hospital Center, Washington, D.C., USA).
Funding
The study was funded by BIOTRONIK AG, Buelach, Switzerland.
Conflict of interest statement
H.M. Garcia-Garcia and R. Waksman were core laboratory members, the remaining authors were investigators of the trial. M. Haude reports grants/contracts from Biotronik, Cardiac Dimensions, OrbusNeich, and Philips; consulting fees from Biotronik, Cardiac Dimensions, Shockwave Medical, and OrbusNeich; honoraria/speaker fees from Biotronik, Cardiac Dimensions, Shockwave Medical, OrbusNeich, and Philips; support to attend meetings/travel support from Biotronik; is a steering committee member of the BIOSOLVE and BIOMAG trials; and is a past president of EAPCI. J. Torzewski reports grants and contracts from Abbott paid to his institution; speaker honoraria and support for attending meetings from Biotronik; and is an associate editor of Cardiovascular Biologics and Regenerative Medicine and Frontiers in Cardiovascular Medicine. J. Escaned reports personal fees/speaker honoraria from Abbott, Boston Scientific, Philips, and Shockwave; has patents from Shared Medical; and is on the advisory boards of Abbott and Philips. J. Iglesias’ institution receives grants or contracts from Terumo, Biosensors, Concept Medical, Biotronik, Abbott Vascular, and Philips/Volcano. J. Iglesias reports consulting fees from Biotronik, Medtronic, Cordis, Terumo, and ReCor Medical; speaker fees/honoraria from Terumo Corp, Biosensors, MedAlliance, OrbusNeich, Concept Medical, Bristol-Myers Squibb/Pfizer, Novartis, Cordis, AstraZeneca, and Philips/Volcano; and support to attend meetings from Biotronik and Amgen. J. Bennett’s institution receives grants or contracts from Shockwave IVLS. J. Bennett receives consulting fees from Biotronik AG and Boston Scientific; speaker fees/honoraria from Biotronik AG, Boston Scientific, and Abbott Vascular; participates in the DSMB of Boston Scientific; and has a leadership or fiduciary role for Biotronik. G. Toth reports consulting fees from Biotronik, Medtronic, Abbott, and Terumo; and honoraria from Biotronik, Medtronic, Abbott Vascular, and Terumo. M. Joner reports grant support from Boston Scientific, Cardiac Dimensions, Edwards Lifesciences, and Infraredx; consulting fees from Alchimedics SAS, Biotronik, TriCares, Veryan, and Shockwave; speaker fees/honoraria from Abbott Vascular, Biotronik, Boston Scientific, Edwards Lifesciences, Cardiac Dimensions, AstraZeneca, ReCor Medical, and Shockwave; travel support from SIS Medical, Edwards Lifesciences, Boston Scientific, and Cardiac Dimensions; and participation in the steering committees of Biotronik and Edwards Lifesciences. R. Toelg reports lecture fees from Biotronik. M. Wiemer reports speaker honoraria and conference attendance support from Biotronik. G. Olivecrona reports lecturer honoraria from Abbott Vascular, Biotronik, and Cordis; is a DSMB member of the SCIENCE trial; and a CEC member of the BIOFREEDOM STEMI trial. H.M. Garcia-Garcia has grants or contracts from Medtronic, Biotronik, Abbott, Neovasc, Corflow, Alucentbio, Philips, and Chiesi (paid to the institution); received consulting fees from Boston Scientific and ACIST; and participates in the DSMB/advisory board of the VIVID study. R. Waksman has grants or contracts from Amgen, Biotronik, Boston Scientific, Medtronic, and Philips IGT; received consulting fees from Abbott Vascular, Biotronik, Boston Scientific, Cordis, Medtronic, Philips IGT, Pi-Cardia, Swiss Interventional Systems/SIS Medical AG, Transmural Systems Inc, and Venous MedTech; received honoraria from AstraZeneca; participates in DSMB/advisory boards of Abbott Vascular, Boston Scientific, Medtronic, Philips IGT, and Pi-Cardia; and is an investor in MedAlliance and Transmural Systems, Inc. The other authors have no conflicts of interest to declare.
Supplementary data
To read the full content of this article, please download the PDF.
