
Abstract
Aims: Bioresorbable scaffolds were designed to overcome the limitations of permanent stents. In the BIOSOLVE-I study we aimed to assess the long-term safety and performance of a drug-eluting absorbable metal scaffold (DREAMS) at three years.
Methods and results: In this prospective, multicentre first-in-man study, 46 patients with 47 de novo lesions were enrolled. We report the final results at three-year follow-up. Mean age was 65.3±9.7 years, lesions were 2.73±0.48 mm in diameter and 10.99±4.59 mm long. Follow-up at three years was available for 44 patients (one patient died of a non-cardiac cause and one patient withdrew consent). Three target lesion failures (TLF) occurred (6.6%), consisting of two clinically driven target lesion revascularisations at scheduled six-month angiography (4.3%) and one myocardial infarction after drug-eluting balloon treatment in a non-target lesion but target vessel at 12-month angiography (2.2%). No cardiac death or scaffold thrombosis occurred. Seven patients had additional angiographic follow-up at 28±4 months: in-scaffold late lumen loss had improved from 0.51±0.46 mm (median 0.28 mm) at 12 months to 0.32±0.32 mm (median 0.20 mm).
Conclusions: The BIOSOLVE-I study showed excellent long-term outcomes at three years with a low TLF rate and no cardiac death or scaffold thrombosis. No TLF event was observed beyond 377 days. (ClinicalTrials.gov Identifier: NCT01168830)
Abbreviations
DES: drug-eluting stent
DREAMS: drug (paclitaxel)-eluting absorbable metal scaffold
IVUS: intravascular ultrasound
LLL: late lumen loss
OCT: optical coherence tomography
PCI: percutaneous coronary intervention
PLGA: polylactic-co-glycolic acid
QCA: quantitative coronary angiography
TLF: target lesion failure
TLR: target lesion revascularisation
Introduction
Drug-eluting stents (DES) have reduced the risk of restenosis, but have limitations such as hypersensitive reaction that interferes with endothelialisation, resulting in late persistent or acquired malapposition, which consequently may lead to late and very late stent thrombosis1. The next evolution in percutaneous coronary intervention (PCI) was the development of bioresorbable scaffolds, which were designed to overcome these limitations of permanent stents1. Additionally, the absence of a rigid metallic cage allows restoration of vessel vasomotion and late expansive remodelling and should not limit future treatment options1,2.
Recent randomised trials comparing bioresorbable scaffolds with permanent DES have shown similar outcomes up to one year3,4. While long-term data from randomised controlled trials, which could potentially show the long-term benefit of bioresorbable scaffolds, are currently pending, long-term data of single-arm ABSORB studies have shown encouraging results with sustained good outcomes up to five years5,6.
The BIOSOLVE-I study was conducted to assess the safety and performance of the drug-eluting absorbable metal scaffold (DREAMS). One-year outcomes have been published previously7. We now aim to assess the long-term safety and performance at three-year follow-up.
Methods
STUDY DESIGN AND POPULATION
The design of the study has been previously described2,7. In brief, the BIOSOLVE-I first-in-man study is a prospective, multicentre study to assess the safety and efficacy of a drug (paclitaxel)-eluting absorbable metal (magnesium-based) scaffold (DREAMS; Biotronik AG, Bülach, Switzerland) in subjects with single de novo coronary artery lesions in a maximum of two separate coronary arteries.
A full list of inclusion and exclusion criteria is displayed at ClinicalTrials.gov (NCT01168830). Patients were eligible if they presented with stable or unstable angina or documented silent ischaemia. A maximum of two single de novo lesions in two separate coronary arteries with reference vessel diameters between 3.0 and 3.5 mm, lesion length ≤12 mm and diameter stenosis between 50% and 99% were allowed. The study was conducted in accordance with the Declaration of Helsinki, good clinical practice, ISO14155, and was approved by the institutional ethics committees at the participating institutions. All patients gave written informed consent prior to any study procedure. Completeness and quality of data were assured by 100% source document verification, adjudication of all serious adverse events by an independent clinical events committee and review of safety data by an independent data safety monitoring board. Independent core laboratories performed the quantitative coronary angiography (QCA), intravascular ultrasound (IVUS) and vasomotion analysis (Medstar Health Research Institute, Washington DC, USA), as well as the optical coherence tomography analysis (Rome Heart Research, Italy).
STUDY DEVICE
DREAMS consists of a balloon-expandable bioresorbable metal scaffold pre-mounted on a rapid exchange delivery system. The scaffold is made from a magnesium alloy. After implantation, the scaffold is gradually absorbed by the surrounding tissue. The scaffold was available in a single length of 16 mm and diameters of 3.25 and 3.5 mm. The scaffold surface is completely coated by a matrix consisting of the carrier polylactic-co-glycolic acid (PLGA) and the active substance paclitaxel. The nominal drug content on the DREAMS is 0.07 µg/mm², with a maximum amount of 8.5 µg of drug on the largest scaffold (![]() 3.5×16 mm).
3.5×16 mm).
The delivery system is a rapid exchange catheter. To facilitate fluoroscopic visualisation and positioning, the pre-mounted scaffold is centred between two radiopaque balloon markers. Both scaffold ends are covered and protected by a scaffold fixation system which prevents accidental dislodgement of the scaffold from the delivery system. The scaffold system is compatible with guidewires of 0.014” (0.36 mm) diameter and guiding catheters with an inner diameter of at least 0.070” (1.78 mm).
Definitions
The primary endpoint of the study was target lesion failure (TLF) at six- and 12-month follow-up. TLF was defined as a composite of cardiac death, target vessel myocardial infarction, and clinically driven target lesion revascularisation (TLR). Secondary endpoints up to one year were previously reported7. Further secondary endpoints were scaffold thrombosis rate and cumulative rates of TLF at 24 and 36 months. Cardiac death, clinically driven TLR and scaffold thrombosis were defined according to the Academic Research Consortium definitions8, and myocardial infarction according to the universal definition of myocardial infarction9.
Procedure
DREAMS was implanted after mandatory predilatation. It was recommended to choose a balloon with a diameter that was 0.5 mm smaller than the reference vessel diameter and was shorter or was the same length as the lesion. When implanting the DREAMS, the inflation pressure had to be maintained for at least 20 seconds for full expansion of the scaffold. Post-dilatation was performed in case of incomplete expansion of the scaffold or residual stenosis. Post intervention, patients were recommended to take at least 75 mg aspirin lifelong and dual antiplatelet therapy for a minimum of 12 months.
Assessments
Clinical follow-up was scheduled at one, six, 12, 24 and 36 months. Patients were consecutively assigned to a mandated six-month (cohort 1) or 12-month (cohort 2) angiographic and IVUS follow-up, but voluntary image follow-up was allowed to be carried out vice versa as well as optical coherence tomography (OCT) being offered to patients on a voluntary basis. No imaging was mandated beyond 12 months. Angiographic, IVUS and OCT assessments were described previously2,7 and were carried out in accordance with established and validated methods10.
Statistical analysis
Data are presented using descriptive statistical methods. For continuous variables, the mean and standard deviation were calculated. For categorical data, absolute and relative frequencies were determined. Interquartile ranges and 95% confidence intervals were calculated when appropriate. Clinical endpoints were summarised using Kaplan-Meier estimates. All statistical analyses were carried out using SAS 9.3 (SAS Institute Inc., Cary, NC, USA).
Results
The study was conducted at five centres in Europe, with 46 patients with 47 lesions being enrolled from July to December 2010. One patient withdrew consent during the course of the study and one patient died of a non-cardiac cause. Further details are provided in Figure 1.
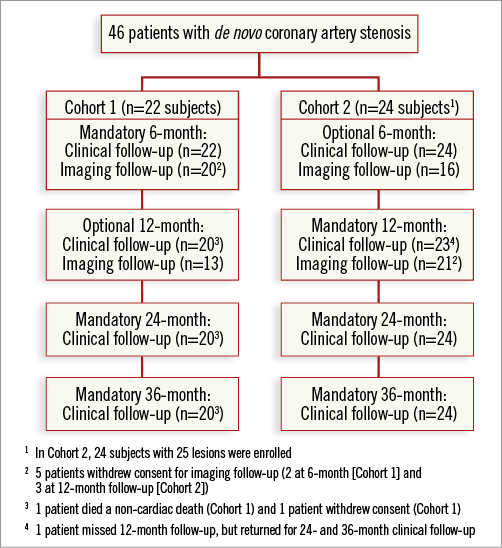
Figure 1. Subject disposition.
Baseline demographics and medical history are displayed in Table 1. The mean age was 65.3±9.7 years (ranging from 42 to 77 years), 58.7% (27/46) of the patients had prior coronary interventions, 32.6% (15/46) had prior myocardial infarctions, and 15.2% (7/46) diabetes mellitus. Mean lesion length was 10.99±4.59 mm and the mean reference vessel diameter was 2.73±0.48 mm. The majority of lesions were type B1 lesions (65.9%, 31/47), followed by type A lesions (25.5%, 12/47) and type C lesions (8.5%, 4/47). Half of the lesions were treated with a 3.25 mm stent (48.9%, 23/47) and half of the lesions with a 3.5 mm stent (24/47). Post-dilatation was performed in 14.9% (7/47). During the procedures, all devices were successfully delivered, resulting in a device and procedural success rate of 100%. Due to a prolonged IVUS investigation, a flow-limiting situation occurred in one patient requiring the use of a second scaffold.
Kaplan-Meier estimates were 6.6% (n=3, [95% CI: 2.2-19.0]) for TLF, a composite of cardiac death, target vessel myocardial infarction and clinically driven TLR, 2.2% (n=1, [95% CI: 0.3-14.7]) for myocardial infarction, and 4.3% (n=2, [95% CI: 1.1-16.3]) for clinically driven TLR. There was no cardiac death and no additional TLF event beyond 377 days. Of the two subjects who underwent a clinically driven TLR at six-month follow-up, one subject was suffering from unstable angina and had an in-scaffold diameter stenosis of 34.5% at six-month follow-up. The second subject received an additional DREAMS in the target lesion during the initial procedure due to a flow-limiting bail-out situation, had stable angina class III and a 53.9% diameter stenosis at follow-up. The third target lesion failure was a myocardial infarction, which occurred at 377 days post index procedure during scheduled 12-month angiography. This subject received two DREAMS during the index procedure, one in the left anterior descending and a second in the obtuse marginal. During the 12-month control angiography, an ostial lesion in the circumflex artery was treated with a DES, and a stenosis of the circumflex at the bifurcation to the obtuse marginal was treated with a drug-eluting balloon. This subject developed a post-interventional rise of ischaemia markers. As, by definition, the target vessel also includes side branches, this myocardial infarction was classified as target vessel myocardial infarction, even though it did not occur in the originally treated obtuse marginal and the DREAMS scaffold had an open lumen with an in-scaffold diameter stenosis of 31.5%. No additional myocardial infarction occurred in non-target vessels. Overall, no cardiac death or scaffold thrombosis occurred.
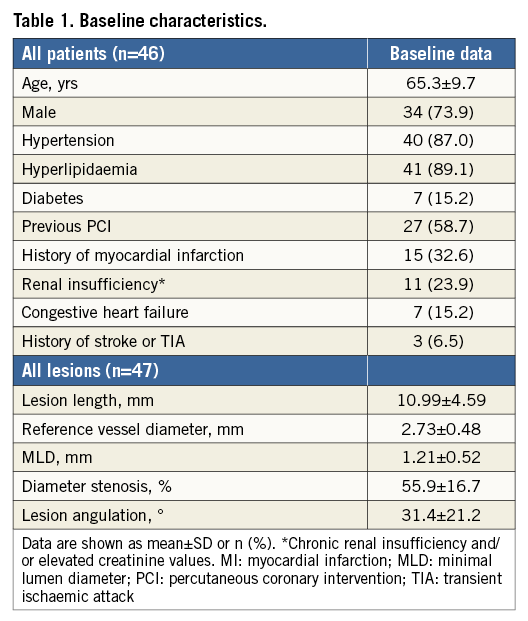
Figure 2 shows a serial imaging assessment of the first patient enrolled. Minimal lumen diameter decreased at six months, followed by a subsequent increase without documentation of malapposed struts up to two years. The binary angiographic stenosis of 54.6% at six months improved at 12 months and remained stable up to 24 months. A focal ectatic lumen was detected at the implantation site by angiography and optical coherence tomography at six months, which disappeared at 12-month control. All struts were covered at six months.
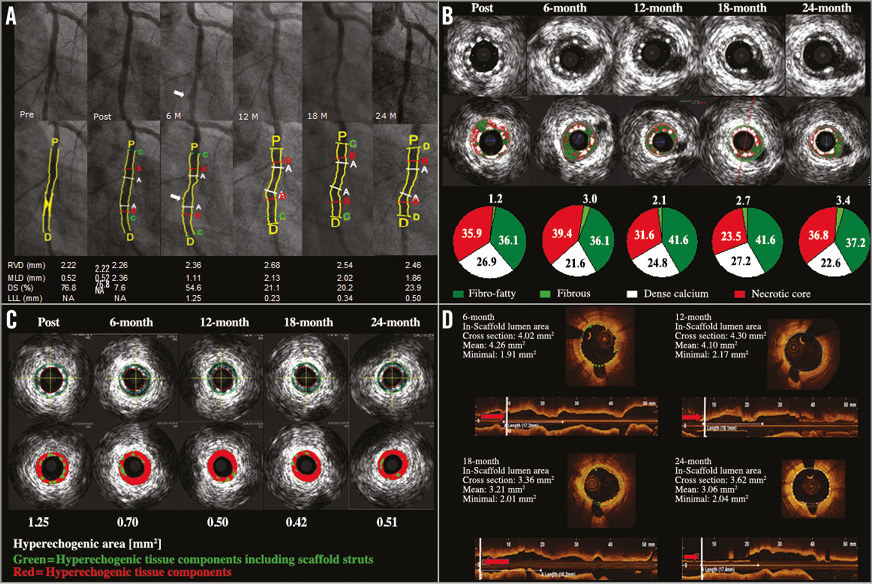
Figure 2. Serial quantitative angiographic, IVUS and OCT assessment of the first patient enrolled in BIOSOLVE-I. Target lesion was the circumflex artery (type B1, length 11.75 mm). Predilatation with 2.5×12 mm compliant balloon (16 atm, two inflations), thereafter implantation with DREAMS scaffold (3.25×16 mm) at 14 atm for 10 seconds. No post-dilatation was required. Clinically, the patient was stable without angina up to 18 months, when a silent ischaemia due to a non-target vessel restenosis in the left anterior descending occurred. After successful treatment with an everolimus-eluting stent, the patient was stable again without any signs of angina. A) Quantitative angiographic assessment of the target lesion in the circumflex coronary artery (A: lesion; B: scaffold; C: in-segment; P: proximal; D: distal). At six months, a small ectasia (arrow) can be detected which disappeared at 18 months. QCA analysis demonstrates the increase in minimal lumen diameter between six and 12 months, which then remained stable until 24-month follow-up. DS: diameter stenosis; LLL: late lumen loss; MLD: minimal lumen diameter; RVD: reference vessel diameter. B) Serial assessment of the greyscale IVUS and matching virtual histology (VH) data. Greyscale IVUS shows excellent apposition with struts covering the side branch post-procedure being absorbed at follow-up. VH data demonstrate a decrease in dense calcium (white) between post-procedure and six months, which remained stable at late follow-up, illustrating the absorption process. C) Echogenicity evaluation of this particular patient demonstrates a large decrease of the hyperechogenic area, and thus the stent struts, within the first six months, followed by a lower decrease between six and 12 months, after which it remains stable until 24-month follow-up. D) OCT of the circumflex coronary artery (same cross-sectional area) at six, 12, 18 and 24 months after DREAMS implantation showing good apposition to the vessel wall. At six months, a small dissection was visible, which disappeared at 12 months. At six months, all struts are covered, with struts beginning to fade. At 12, 18, and 24 months, remnants of the struts are hardly discernible but a shadowing behind the remnants is still visible.
An angiographic assessment after 12 months was performed in seven patients. Mean follow-up was 28±4 months. The minimal in-scaffold lumen diameter was 2.59±0.37 mm post procedure, decreased to 2.08±0.53 mm at 12 months, and increased to 2.27±0.49 mm at 28 months. In-segment minimal lumen diameter was 2.31±0.47 mm, 2.04±0.52 mm and 2.21±0.55 mm, respectively. Similarly, in-scaffold and in-segment late lumen loss were higher at 12 months (mean in-scaffold LLL of 0.51±0.46 mm, median 0.28 mm, mean in-segment LLL of 0.28±0.34 mm, median 0.26 mm) and lower at long-term follow-up (mean in-scaffold LLL of 0.32±0.32 mm, median 0.20 mm, mean in-segment LLL of 0.11±0.18 mm, median 0.13 mm) (Figure 3).
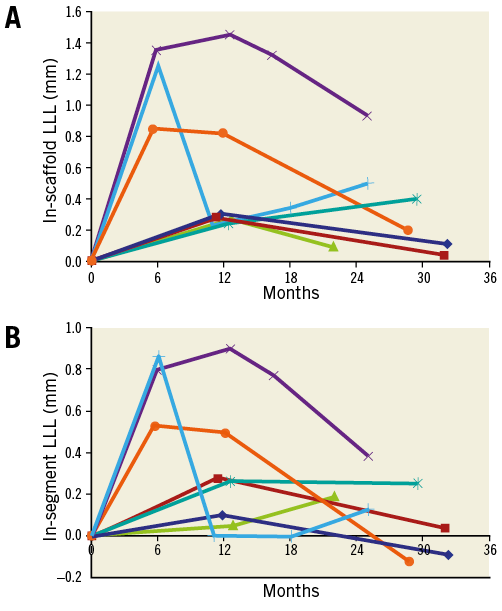
Figure 3. In-scaffold and in-segment LLL of seven patients with angiographic QCA assessment up to three years. A) In-scaffold LLL. B) In-segment LLL. Each line reflects the course of LLL per patient. The light blue line with “+” reflects data of the patient displayed in Figure 2. LLL increased from post-procedure, was highest around six and 12 months, and then decreased. At a mean follow-up of 28±4 months, mean in-scaffold LLL was 0.32±0.32 mm (median 0.20 mm) and in-segment LLL 0.11±0.18 mm (median 0.13 mm). LLL: late lumen loss
Greyscale IVUS assessments up to 18 months were available for four patients from one centre and remained stable throughout follow-up (Table 2). Virtual histology was available in three patients (Figure 4).
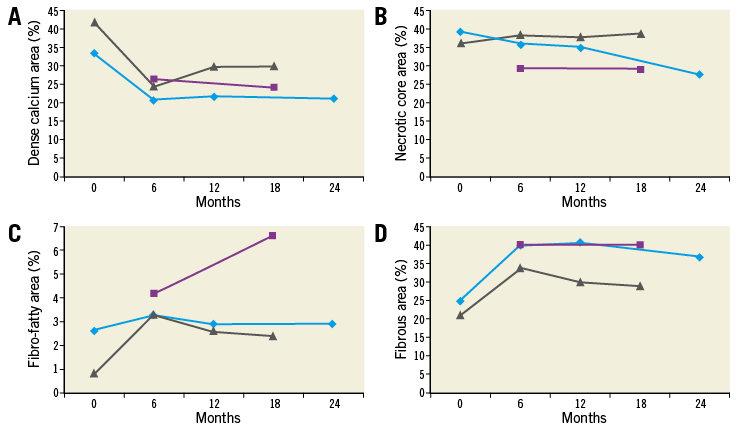
Figure 4. Virtual histology of three patients with assessments beyond 12 months. A) Dense calcium. B) Necrotic core area. C) Fibro-fatty area. D) Fibrous area [%]. Each line reflects the course per patient. The light blue line reflects data of the patient displayed in Figure 2 and Figure 3. The purple line reflects the data of the patient with a purple line in Figure 3.
Discussion
We report the first long-term outcomes of a drug-eluting magnesium-based bioresorbable metallic scaffold up to three years. The main findings are that DREAMS showed an excellent safety profile up to three years with no TLF beyond 377 days and no cardiac death or scaffold thrombosis. Specifically, two TLRs occurred in cohort 1 during scheduled six-month angiography and one myocardial infarction in cohort 2 during scheduled 12-month imaging assessment, resulting in a TLF rate of 6.6%. Both TLR patients were symptomatic, but had a relatively low diameter stenosis of 34.5% and 53.9%, respectively. The decision to perform a revascularisation was operator-driven. The myocardial infarction was related to a non-target lesion intervention and was not associated with the scaffold itself. One could speculate that the full strut coverage at six months and the high rate of apposed struts2,7 might have influenced the low rate of myocardial infarction and the absence of scaffold thrombosis in BIOSOLVE-I.
For angiographic parameters, it has been shown that they remain stable if not improve over time. In eight patients of the PROGRESS study with implantation of the bare absorbable metal scaffold (AMS), a serial angiographic and IVUS assessment with additional follow-up between 12 and 28 months was performed. Compared to four months, this imaging follow-up revealed a reduction in neointima, LLL (-0.23 mm, range -0.93 to 0.05) and diameter stenosis (8%), and an increase in scaffold cross-sectional area, and MLD (0.22 mm, one lesion was unchanged, the remaining lesions improved). The authors concluded that, after completion of degradation, there was a stability and durability in angiographic and IVUS performance parameters, and that there were no adverse effects to the vessel wall at the site of AMS implantation11.
Similarly, the BIOSOLVE-I study showed stable angiographic outcomes between six and 12 months, with an in-scaffold LLL of 0.65±0.50 mm at six months and 0.52±0.39 mm at 12 months7. Additionally, in seven patients of BIOSOLVE-I who had an additional angiographic follow-up beyond 12 months, angiographic parameters continued to improve; it appears that late lumen enlargement is higher in lesions with high late lumen loss at six- to 12-month follow-up. While the total number of seven patients allows only limited conclusions, it is still remarkable that, despite the high initial LLL, the median in-scaffold LLL at 28 months was reduced to 0.20 mm (interquartile range 0.11-0.40 mm, mean 0.32±0.32 mm) and hence is similar to the eight-month median in-scaffold LLL of 0.18 mm reported for new-generation DES in a recent meta-analysis of the ESC Task Force12. This may explain the favourable long-term outcomes with no additional TLR beyond six-month follow-up.
In ABSORB cohort B patients, it has been shown that LLL remains stable between one and three years (in-scaffold LLL of 0.27±0.34 mm at one year and 0.29±0.43 mm at three years)6, and, in a small series of 14 patients enrolled in ABSORB cohort A, late lumen enlargement between two and five years was observed13. This might correspond to our findings inasmuch as (a) we observed late lumen enlargement mainly in patients with a high LLL at six to 12 months, whereas the mean LLL in ABSORB cohort B was relatively small, and (b) the Absorb BVS (Abbott Vascular, Santa Clara, CA, USA) has a longer scaffolding time than DREAMS, so that late lumen enlargement may be postponed.
We deem the long-term results of BIOSOLVE-I valuable as they show the results of the absorption process of DREAMS over time. Specifically, with the limitation of only seven patients assessed, it has been shown that long-term angiographic performance criteria improve over time to values comparable with new-generation DES. Recently, six-month data of the second-generation DREAMS (DREAMS 2G) have been published15, which showed a marked reduction in in-segment and in-scaffold LLL when compared to first-generation DREAMS and which hopefully will also show continuous lumen enlargement over time.
Limitations
As with all first-in-man studies, BIOSOLVE-I was not randomised, had patients with low clinical and anatomical complexity, and included a small number of patients. This resulted in wide confidence intervals and hampers comparison to other stents. Still, the study served its intention to assess the safety and performance of the device. The high follow-up compliance at three years with information in all but one patient who withdrew consent is a strength of the study. Considering the excellent clinical outcomes, and the improvement of angiographic parameters such as LLL observed in a small number of patients assessed beyond 12 months, it would be of interest to see angiographic, IVUS and OCT assessments several years after implantation in all patients. Future studies of bioresorbable scaffolds should include a serial imaging follow-up up to three or even five years to study the process of device absorption over time and to determine the validity of LLL assessment at six or 12 months for predicting long-term outcomes.
Conclusion
Despite suboptimal angiographic results at six and 12 months, the BIOSOLVE-I study showed good long-term outcomes in de novo lesions with a TLF rate of 6.6% at three years and no cardiac death or scaffold thrombosis which is comparable to other competing bioresorbable scaffolds. No TLF event was observed beyond 377 days.
| Impact on daily practice This report presents the first long-term outcome of a drug-eluting bioresorbable metal scaffold with a TLF rate of only 6.6% at three years and no scaffold thrombosis. These results suggest the applicability of this scaffold for clinical use in the patient and lesion scenarios outlined in the BIOSOLVE-I trial. |
Acknowledgements
We thank Beatrix Doerr for her expert medical writing assistance.
Funding
This study was funded by Biotronik AG, Bülach, Switzerland.
Conflict of interest statement
M. Haude reports receiving study grants and lecture fees from Biotronik, Abbott Vascular, Cardiac Dimensions, Medtronic, Volcano, and Lilly. R. Waksman reports personal fees from Biotronik, Medtronic, Abbott Vascular, grants and personal fees from AstraZeneca, Boston Scientific, and grants from The Medicines Company and Edwards Lifesciences. J. Koolen reports receiving lecturer and consultancy fees from Medtronic and Biotronik. The other authors have no conflicts of interest to declare.
