Abstract
Aims: Although the proof of concept of the bioresorbable vascular scaffold (BRS) is well documented, device-related adverse outcomes with first-generation BRS indicate longer-term surveillance. The current study provides insights into the safety and performance of the MeRes100, a novel second-generation sirolimus-eluting BRS, beyond one-year up to three-year follow-up (FU).
Methods and results: A total of 108 enrolled patients with de novo coronary artery lesions who underwent implantation of MeRes100 as part of the first-in-human MeRes-1 trial were followed up clinically beyond one year at two and three years and with multiple modality imaging at six months and two years. At three-year FU, the cumulative major adverse cardiac events rate was 1.87%, in the form of two ischaemia-driven target lesion revascularisations. No scaffold thrombosis was reported. Between six months and two years at quantitative coronary angiography, in-segment late lumen loss (LLL) (0.15±0.22 mm vs. 0.23±0.32 mm; p=0.18) and in-scaffold LLL (0.13±0.22 mm vs. 0.24±0.34 mm; p=0.10) changed insignificantly. IVUS subset analysis revealed a non-significant reduction in mean lumen area (6.17±1.28 mm2 vs. 5.47±1.50 mm2; p=0.21) and minimum lumen area (5.14±1.19 mm2 vs. 4.05±1.42 mm2; p=0.10) at two years compared to post-procedural measurements. OCT subset analysis demonstrated 99.24±2.27% neointimal strut coverage.
Conclusions: The extended outcomes of the MeRes-1 trial demonstrated sustained efficacy and safety of the MeRes100 BRS with maintained lumen patency up to two years by multimodality imaging and no very late scaffold thrombosis up to three-year clinical FU.The MeRes-1 trial is registered at the Clinical Trials Registry-India. CTRI Number: CTRI/2015/04/005706
Introduction
Percutaneous coronary intervention (PCI) with implantation of an advanced drug-eluting stent (DES) is currently the cornerstone of treatment for obstructive coronary artery disease1. However, the presence of a permanent metallic implant in the coronary arteries also creates an ongoing risk of occurrence of very late adverse cardiac events at a rate of 2-3% per year up to five years and even beyond2,3. It is this limitation of DESs which gave rise to the concept of a temporary implant with drug elution for acute benefit and its eventual disappearance to eliminate shortcomings due to the permanently caged metallic vessel. This led to the development of the bioresorbable vascular scaffold (BRS). Following its introduction in 2010, use of the first-generation BRS (Absorb™; Abbott Vascular, Santa Clara, CA, USA) proliferated in the initial years.
However, subsequent randomised trials demonstrated that, while the BRS was as effective as best in class DES, when compared to an everolimus-eluting stent up to three-year follow-up it was less safe and associated with an increased device thrombosis at all time points4. Some of the potential reasons for these increased thrombogenicity outcomes included thicker struts (>150 µm) resulting in laminar flow disruptions, delayed endothelialisation, unfavourable dismantling during the resorption process, delayed resorption and also technique-related inefficiencies leading to inadequate implantation and incomplete scaffold apposition5. It was therefore hypothesised that many of the limitations of first-generation BRS could be overcome by a thin-strut user-friendly BRS, which would also be less thrombogenic at all time points (early, late and very late).
The MeRes100™ (Meril Life Sciences Pvt. Ltd., Vapi, India) is a novel thin-strut second-generation sirolimus-eluting poly-L-lactic acid (PLLA)-based bioresorbable coronary scaffold. The first-in-human MeRes-1 trial demonstrated the MeRes100 BRS to be safe and effective for the treatment of de novo coronary lesions as evidenced by the low major adverse cardiac events (MACE) rate (0.93%) at one-year follow-up. Importantly, none of the patients experienced scaffold thrombosis at one-year follow-up. Vascular imaging using quantitative coronary angiography (QCA), intravascular ultrasound (IVUS) and optical coherence tomography (OCT) also confirmed favourable vascular response during six-month follow-up6. As very late cardiac events related to scaffold thrombosis beyond one year and up to three years have been of great concern with the first-generation BRS7,8,9,10,11, we analysed three-year clinical outcomes of the second-generation BRS, the MeRes100. Moreover, the results of multiple imaging modalities (QCA, IVUS, and OCT) at two-year follow-up were also studied.
Methods
TRIAL DESIGN
MeRes-1 was a prospective, multicentre, single-arm, open-label clinical trial which enrolled 108 patients from 13 participating centres across India between May 2015 and April 2016 (Clinical Trials Registry-India: CTRI/2015/04/005706). The inclusion and exclusion criteria, study endpoints and definitions have been published previously6. The study complied with the ICH-GCP guidelines, the Declaration of Helsinki, ISO 14155, and was approved by the local ethics committees at all participating institutions. All enrolled patients (intention-to-treat population) gave informed consent for trial participation.
STUDY DEVICE
The design and specifications of the MeRes100 BRS (Figure 1) have been described elsewhere6. In brief, the MeRes100 BRS is a balloon-expandable PLLA polymer backbone scaffold with a strut thickness of 100 µm, coated with a bioresorbable matrix for controlled release of an antiproliferative drug (sirolimus, 1.25 μg/mm2). The degradation of the scaffold is expected to occur within two to three years of implantation. The presence of three pairs of platinum radiopaque markers that are 120º apart from each other at either end of the scaffold facilitates angiographic placement. Currently, the MeRes100 BRS scaffold is available in diameters of 2.25-4.50 mm and lengths of 13-48 mm.
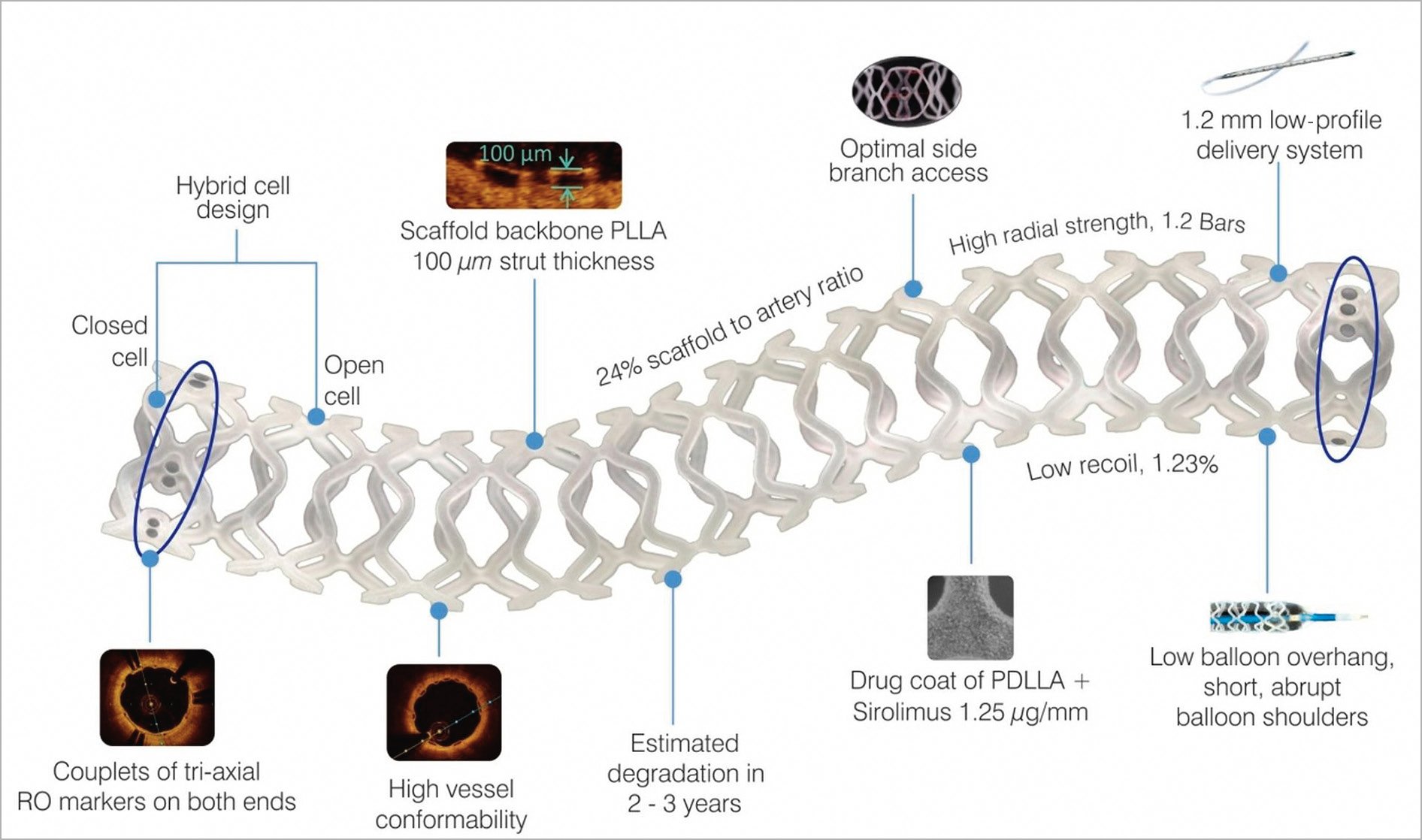
Figure 1. MeRes100 BRS design.
IMPLANTATION TECHNIQUE
PCI was performed according to the standard guidelines to treat target lesions; these have been published previously6,12. Dual antiplatelet therapy with aspirin (75-150 mg/day) and clopidogrel (75 mg/day) or prasugrel (10 mg/day) or ticagrelor (90 mg/day) was prescribed to all patients for a minimum duration of one year, beyond which a switch to single antiplatelet therapy with aspirin alone was left to the operator’s discretion.
ENDPOINTS AND FOLLOW-UP
The clinical endpoints of the study were MACE (a composite of cardiac death, any myocardial infarction [MI], and ischaemia-driven target lesion revascularisation [ID-TLR]) and scaffold thrombosis at three-year follow-up. All MACE were independently adjudicated by a clinical events committee, and a data safety monitoring board evaluated patient safety. The major imaging endpoints for the present analysis included in-scaffold and in-segment late lumen loss (LLL) by QCA, minimum lumen area and neointimal hyperplasia (NIH) area by OCT, and scaffold and lumen area by IVUS imaging at two-year follow-up. Currently, clinical follow-up is complete up to three years.
MULTIMODALITY IMAGING ASSESSMENTS
QCA was performed in a predefined subset of patients at baseline, six months and two years. Out of these, based on patient consent, IVUS (n=10) and OCT (n=9) were performed at pre-designated sites with technical facilities for OCT and IVUS in different subsets of patients. Hence, both OCT and IVUS were not performed in the same patient. The QCA parameters were analysed at the Cardiovascular Research Centre, Sao Paulo, Brazil, using dedicated QAngio® XA software, version 7.3 (Medis medical imaging systems, Leiden, the Netherlands). The OCT and IVUS images were analysed by an independent core laboratory, Cardialysis BV, Rotterdam, the Netherlands. The OCT and IVUS images were captured using a Fourier-domain OCT system (C7 Dragonfly™ and C7-XR™; St. Jude Medical, St. Paul, MN, USA) and Echoplaque, version 3.0.28 software from INDEC Systems, Inc. (Los Altos, CA, USA), respectively. The details of the procedures have been published elsewhere6.
STATISTICAL ANALYSIS
This is a feasibility study designed to provide preliminary observations and generate hypotheses for future studies. Since there was no hypothesis testing in this trial, the sample size was not calculated based on endpoint hypothesis. However, the sample size requirement was determined by assessing the minimal number of patients required to provide reliable and non-trivial results. Continuous variables were expressed as the mean±standard deviation (SD), and categorical variables were expressed as frequency and percentages. Comparisons of clinical, angiographic or imaging outcomes of patients were performed using either a paired t-test (for normally distributed data) or the Wilcoxon signed-rank test (non-normally distributed data). A p-value <0.05 was considered statistically significant. Statistical analysis was performed using SAS, version 9.3 (SAS Institute, Cary, NC, USA).
Results
The MeRes-1 trial enrolled 108 patients (116 lesions) with a mean age of 50.13±8.81 years. Among included patients, 30 (27.78%) had diabetes mellitus, 45 (41.67%) had hypertension, and 37 (34.26%) had MI. The majority (51.9%) of the patients presented with stable angina. The baseline clinical and lesion characteristics and the study flow chart up to three-year follow-up are presented in Table 1 and Figure 2, respectively.
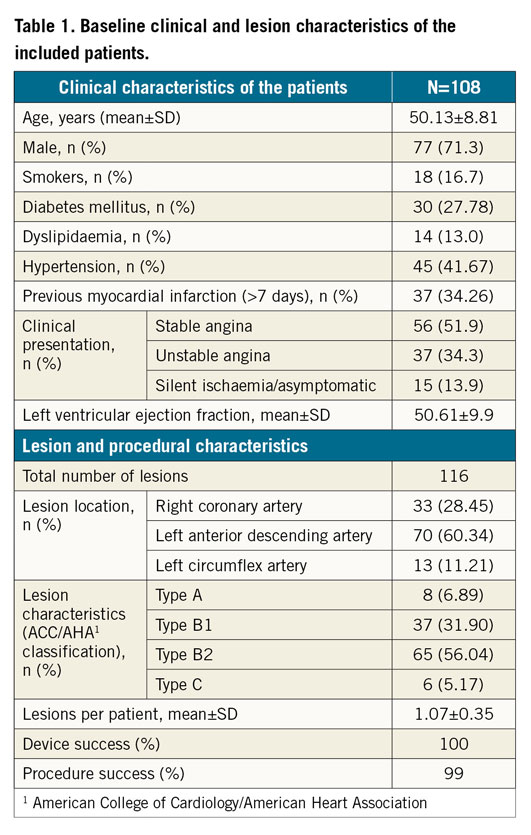
Figure 2. Flow chart of patient follow-up.
Clinical events up to 12 months have been reported previously6. At six-month follow-up, one patient had died due to aminophylline-induced anaphylactic shock (non-cardiac death). Hence, the subsequent follow-up was on 107 patients. The cumulative MACE rate at 12-month follow-up was 0.93% accounted for by a single case of ID-TLR (Table 2). In this case, a 60-year-old female with a history of hypertension was successfully treated with the MeRes100 (2.75×24 mm) for 99% occlusion of the mid right coronary artery. As six-month angiography revealed in-scaffold restenosis (90% occlusion), the patient was treated with a 2.75×28 mm DES with TIMI 2 flow. Between one and three years there was one incidence of ID-TLR. A 45-year-old male with a previous history of smoking and alcohol consumption was diagnosed with 70% stenosis in the proximal left anterior descending artery and impaired left ventricle function. The patient was treated with implantation of the MeRes100 (3.5×19 mm). At the 24-month follow-up visit, scheduled angiography showed 80% in-scaffold restenosis with an LLL of 1.69 mm for which he was treated with implantation of a 3.5×24 mm BioMime™ stent (Meril Life Sciences Pvt. Ltd.). The cumulative MACE rate was found to be 1.87% (n=2/107) at three-year clinical follow-up. None of the patients experienced scaffold thrombosis up to three-year clinical follow-up. There were three (2.80%) target vessel revascularisations (TVR; non-target lesion) at three-year clinical follow-up.
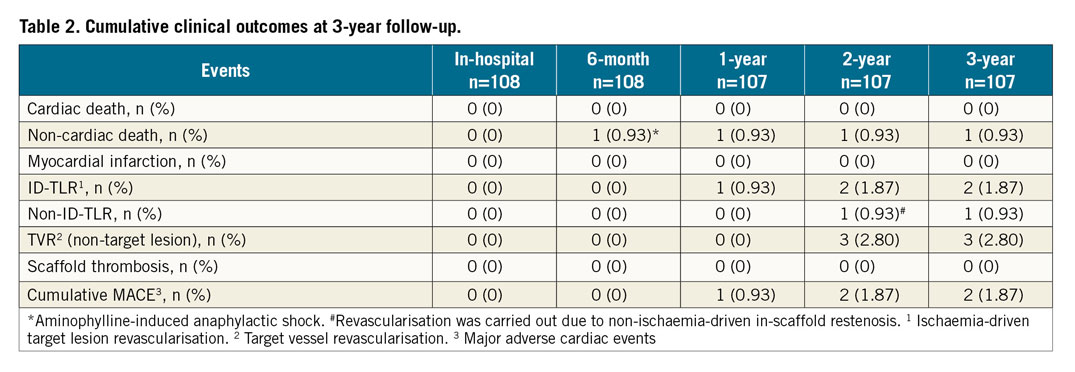
Paired QCA evaluation was performed in a selected cohort of patients (n=29) at baseline, post procedure, six months, and two years (Table 3). In-scaffold LLL (0.13±0.22 mm vs. 0.24±0.34 mm; p=0.10) and in-segment LLL (0.15±0.22 mm vs. 0.23±0.32 mm; p=0.18) at two years did not differ significantly from six-month measurements. QCA examination revealed one in-segment binary restenosis and one in-scaffold restenosis. Figure 3 depicts the cumulative frequency distribution curve for in-segment and in-scaffold LLL at two-year follow-up.
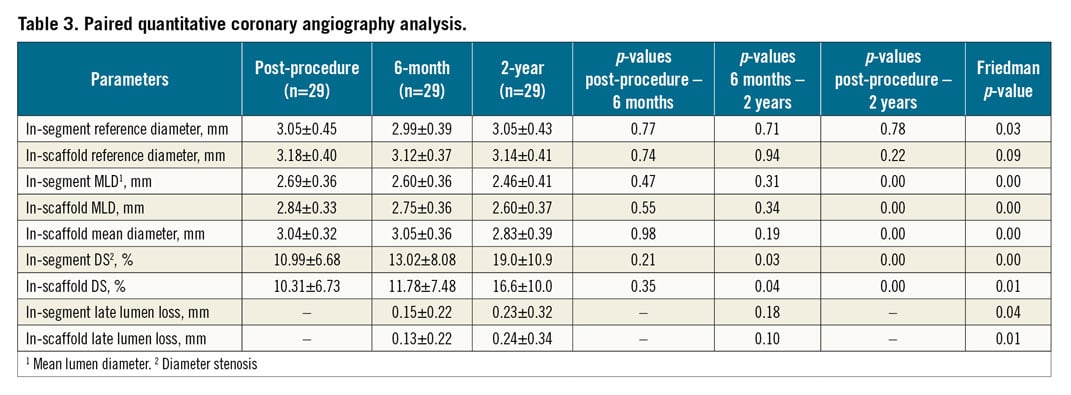
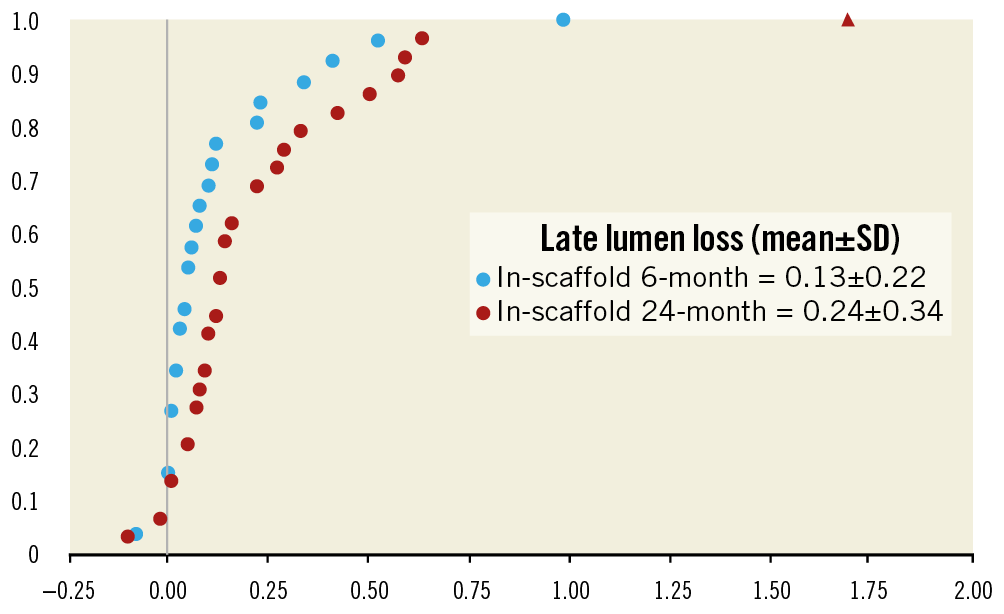
Figure 3. Cumulative frequency distribution curve for in-scaffold late lumen loss at six-month and at two-year follow-up. The triangle symbol represents the 1.69 mm LLL for the patient who underwent an ID-TLR at two-year angiographic follow-up.
Paired analyses for IVUS and OCT were completed in a subset of 10 and nine patients, respectively (Table 4, Table 5). Neointimal coverage of struts was almost complete (99.24±2.27%) with a homogeneous pattern at two years. The number of discernible struts decreased from 185.67±43.77 at post procedure to 87.44±17.26 at two years. Figure 4 illustrates a case with multimodality imaging.
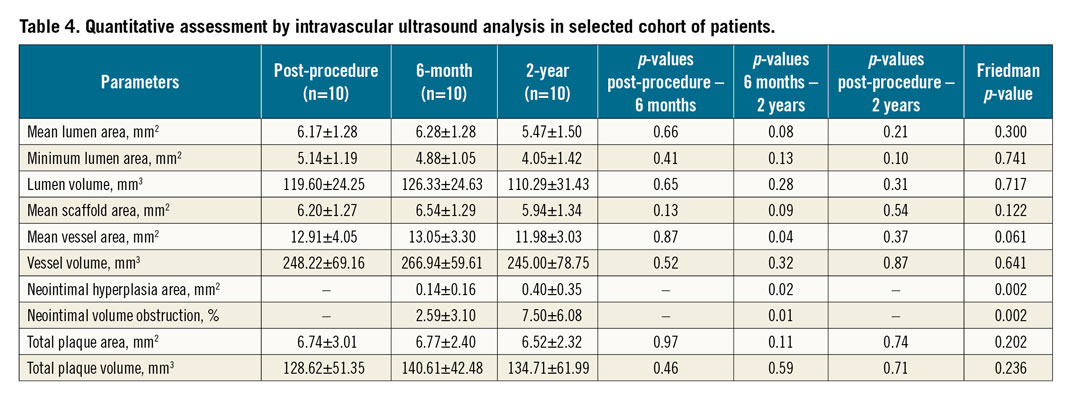
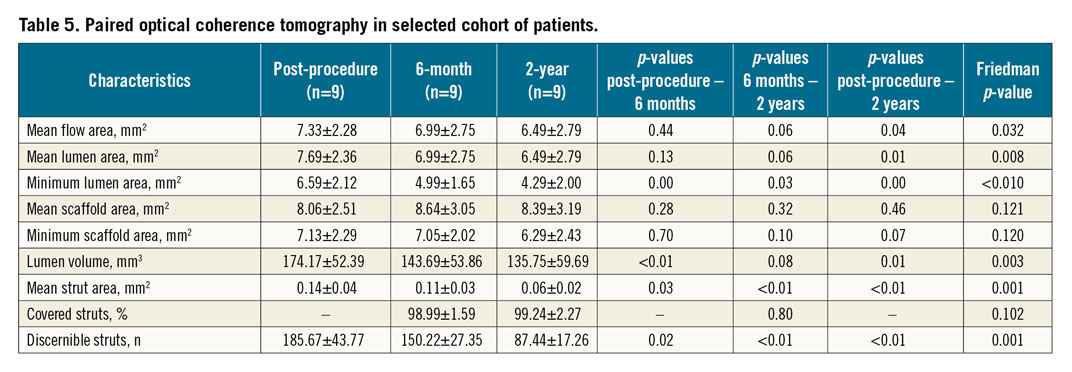
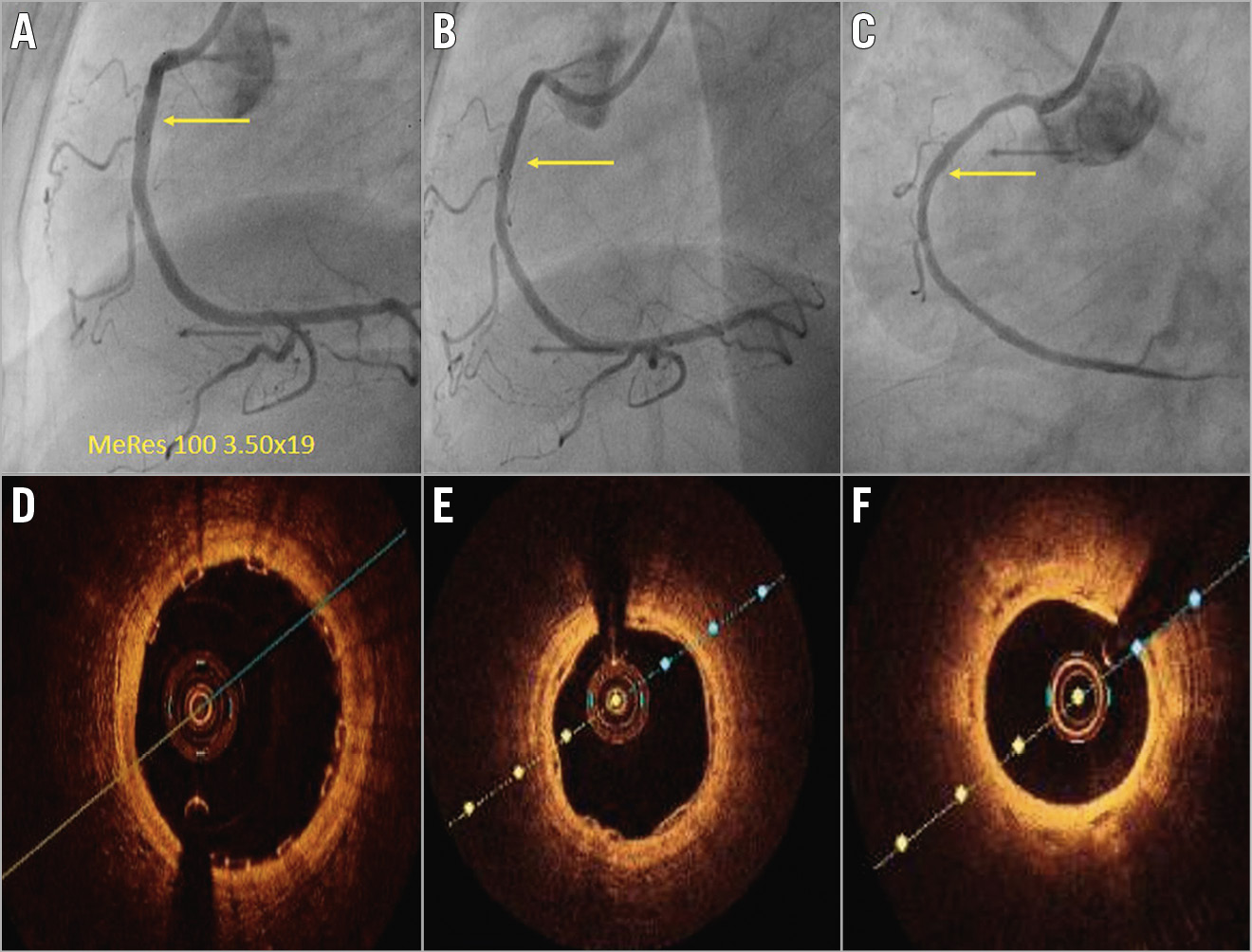
Figure 4. Serial angiographic evaluation at post-procedure, six-month and two-year follow-up (A-C) and OCT analyses at six-month and two-year follow-up (D-F). A) Post-procedural angiographic evaluation confirms successful implantation of 3.5x19 mm MeRes100 in the mid right coronary artery. Follow-up angiographic images at six-month follow-up (B), and at two-year follow-up (C). Matched cross-sectional OCT images of the implanted scaffold after the procedure (D), at six-month follow-up (E), and at two-year follow-up (F).
Discussion
To the best of our knowledge, this is the first trial assessing the safety and performance of second-generation thin-strut BRS with multiple imaging modalities beyond one-year follow-up. The primary findings of the MeRes-1 trial are: 1) good clinical safety as evidenced by the low cumulative MACE rate (1.87%) accounted for by two cases of ID-TLR by three years; 2) no occurrence of very late scaffold thrombosis up to three years; 3) between six months and two years, in-segment LLL (0.15±0.22 mm vs. 0.23±0.32 mm; p=0.18) and in-scaffold LLL (0.13±0.22 mm vs. 0.24±0.34 mm; p=0.10) changed insignificantly with one in-segment binary restenosis and one in-scaffold restenosis; 4) a non-significant reduction in IVUS-measured mean lumen area (6.17±1.28 mm2 vs. 5.47±1.50 mm2; p=0.21), minimum lumen area (5.14±1.19 mm2 vs. 4.05±1.42 mm2; p=0.10) at two years compared to post-procedural measurements; and 5) demonstration of almost complete neointimal strut coverage by OCT at six months (98.99±1.59%) and two years (99.24±2.27%).
A temporary implant which treats the lesion and disappears in two to three years leaving the artery in its natural state without a foreign body is a physiologically logical and institutively attractive option. After resorption of the BRS once the artery is restored to its natural state, presumably there should be freedom from cardiac events, ability to discontinue antiplatelet therapy at will if the need arises, ability to bypass graft these vessels if the need arises in future, etc.13. However, the first-generation BRS had numerous limitations. It was a large-profile bulky device with unique technical characteristics which was difficult to deliver and implant. It also required meticulous attention to the implantation technique and had a learning curve. Hence, its uptake amongst most interventional cardiologists remained low as a niche device. While the BRS was effective, the biggest concern about the first-generation device was its safety14. A meta-analysis of seven randomised trials (n=5,583 patients) concluded that there was a higher incidence of device thrombosis at two-year follow-up with Absorb BRS than with the DES (2.3% vs. 0.7%; relative risk: 3.35 [95% confidence interval: 1.96-5.72; p<0.0001]). Moreover, the analysis showed that very late scaffold thrombosis between one and two years occurred only in the Absorb BRS group2. In vitro and in vivo studies have demonstrated that thicker struts create disturbances of laminar flow and shear stress predisposing to increased thrombosis and the first generation BRS had a strut thickness of 150 μm. Thicker struts also endothelialise late and unevenly. They also resorb later and add bulk to the scaffold, making it difficult to deliver. So, on all counts, decreasing the strut thickness of the BRS could overcome many of the limitations of first-generation BRS and most importantly could make it less thrombogenic and safer15.
The MeRes100 incorporates several important improvements to make it technically user-friendly, including novel architecture (hybrid cell design, closed cells at the edges and open cells along the length), low strut thickness (100 µm), good radiopacity (couplets of tri-axial platinum radiopaque markers fixed circumferentially 120º apart from each other at either end of the scaffold), high radial strength (1.2 bar), high deliverability-flexibility and availability of the scaffold in wider scaffold length or diameter. The MeRes-1 trial demonstrated a low event rate (0.93%), accounted for by a single incidence of ID-TLR at one-year clinical follow-up. Multiple imaging modalities at six-month follow-up in a predefined subset demonstrated (QCA-derived LLL of 0.15±0.22 mm, in-scaffold percentage diameter stenosis of 11.78±7.48, IVUS-derived percentage neointimal volume obstruction of 2.59±3.10, OCT-identified percentage strut coverage of 98.99±1.59) maintained safety and efficacy of the MeRes100 at short-term follow-up. Computed tomography angiography analysis at one year further confirmed vessel patency and no difference in minimum lumen area (IVUS-derived minimum lumen area at six months: 4.88±1.05 mm2 vs. computed tomography angiography-derived minimum lumen area at two years: 4.29±2.00 mm2). Furthermore, three-year clinical outcomes and two-year multiple imaging modality observations also replicate evidence of the safety and performance of the MeRes100. In the MeRes-1 trial, IVUS analysis demonstrated an insignificant reduction in mean lumen area, minimum lumen area, and lumen volume during follow-up. However, OCT analyses revealed a significant decrease in minimum lumen area, mean lumen area, and lumen volume as a result of neointimal proliferation in the implanted area. These contradictory findings could be explained by a smaller sample size in each arm.
The current analysis of the MeRes-1 trial is one of the few to have evaluated the safety and performance of BRS beyond one-year follow-up. Cumulative MACE rates were found to be 1.87% (n=2/107) at three-year follow-up. There was only one ID-TLR between 12 months and three years. No patient experienced scaffold thrombosis up to three-year clinical follow-up. While this could be due to the lower strut thickness of the MeRes100 leading to more rapid endothelialisation as well as resorption and lower thrombogenicity, it could also have been due to the fact that all MeRes100 were implanted using a dedicated implantation technique as learned with the first-generation BRS16,17. To date, long-term clinical follow-up is available for the Absorb BRS and the DESolve® BRS (Elixir Medical Corporation, Milpitas, CA, USA) with concerns regarding long-term safety2,18. The DESolve Nx trial demonstrated a cumulative MACE rate of 7.4% (n=9/122) at two-year clinical follow-up, including three cardiac deaths, one MI and five TLRs. There was only one case of probable scaffold thrombosis which occurred during the first month after implantation18. In contrast to the Absorb BRS, none of the patients in the MeRes-1 trial experienced very late scaffold thrombosis2.
Study limitations
Our study has some noteworthy limitations. The results of our study are based on the first-in-human study in a relatively small number of stable patients with non-complex coronary lesions. Also, the results cannot be generalised to more complex lesions or higher-risk patients. The low MACE rate needs a cautious interpretation given the single-arm trial design without any active comparator. Any inference of non-inferior or superior safety against current-generation drug-eluting stents or scaffolds is not possible. However, encouraging long-term clinical outcomes and multiple modality imaging analysis provide the basis for further studies in a more complex and larger patient population.
Conclusions
The first-in-human MeRes-1 thin-strut scaffold trial demonstrated favourable clinical outcomes in patients with de novo non-complex coronary lesions at three-year follow-up as evidenced by a low rate of adverse events and the absence of scaffold thrombosis. Moreover, multimodality invasive imaging assessment at two years reaffirmed favourable endpoints for safety and efficacy.
|
Impact on daily practice Recent large real-world registry randomised controlled trials as well as meta-analysis of first-generation BRS demonstrated increased thrombogenicity and hence safety concerns. Consequently, more information related to long-term safety and efficacy of available BRS, especially the second-generation thin-strut BRS, is crucial. The present study demonstrated a low rate of adverse events with no scaffold thrombosis up to three years and maintained measures of safety and efficacy on multimodality imaging. These results are encouraging for the use of thin-strut BRS in clinical practice in a selected subgroup of patients until large pivotal and long-term data are available to prove its long-term advantages. |
Acknowledgements
We express our sincere gratitude to Samuel K. Mathew, P.C. Rath, Vijay Trehan, Sanjeev Bhatt, and Imrankhan Lohani, who were invaluable collaborators in the execution of the MeRes-1 trial.
Funding
The MeRes-1 clinical trial was funded by Meril Life Sciences Pvt. Ltd., India.
Conflict of interest statement
A. Seth and A. Abizaid are external scientific advisors to Meril Life Sciences Pvt. Ltd. The other authors have no conflicts of interest to declare. The Guest Editor is a consultant for Edwards Lifesciences.
