
Abstract
Aims: The aim of this study is to compare the SUPRAFLEX sirolimus-eluting stent (SES) with the XIENCE everolimus-eluting stent (EES) with respect to target lesion failure (TLF) at 12 months in a non-inferiority trial in a “real-world” patient population.
Methods and results: This is a prospective, randomised, 1:1 balanced, controlled, single-blind, multicentre study comparing clinical outcomes at 12 months between SUPRAFLEX and XIENCE in an “all-comers” patient population, comprising a total of 1,430 enrolled subjects with symptomatic coronary artery disease who qualify for percutaneous coronary interventions at 23 centres in Europe. The primary endpoint is a non-inferiority comparison of the device-oriented composite endpoint target lesion failure (cardiac death, target vessel myocardial infarction and clinically indicated target lesion revascularisation) of the SUPRAFLEX group to the XIENCE group at 12 months post procedure. Secondary endpoints include the patient-oriented composite endpoint, target vessel failure, mortality, myocardial infarction, revascularisation and stent thrombosis rates (ARC classification).
Conclusions: The TALENT trial aims to assess the safety and effectiveness of the thin-strut SUPRAFLEX compared to the current standard of care (XIENCE EES) in patients with atherosclerotic lesions. This will provide valuable information on the impact of this thin-strut device in an all-comers population.
Introduction
Coronary stent technology has been constantly improving in order to diminish adverse outcomes for patients who undergo percutaneous coronary intervention (PCI). After the 40th anniversary of Andreas Grüntzig’s first angioplasty (using just a balloon), the progress in stent and intravascular device development has maintained a constant pace - from balloon to bare metal stents (BMS), followed by the first-generation drug-eluting stents (DES), and now the biodegradable polymer DES.
Biodegradable polymer was developed to control the drug release temporarily and then dissolve, leaving a bare metal stent-like platform. The efficacy of biodegradable polymer-coated DES has been shown to be non-inferior to durable polymer DES in several studies1,2,3. A recent study showed that biodegradable polymer DES were superior to durable polymer DES with respect to a lower rate of target lesion failure at 12 months4. In addition, a pooled analysis of large multicentre randomised trials evidenced lower rates of target vessel revascularisation and very late stent thrombosis in biodegradable polymer devices compared to those with durable polymers5.
To settle all of these issues, novel stents should focus on incremental improvements in (i) strut thickness and shape of their design, (ii) metallic alloy composition, (iii) drug release kinetics, and (iv) polymer thickness and durability3. The sirolimus-eluting cobalt-chromium coronary stent SUPRAFLEX (Sahajanand Medical Technologies Pvt. Ltd., Surat, India) has a thin strut thickness and a biodegradable polymer technology. In the FLEX Registry, the SUPRAFLEX sirolimus-eluting stent (SES) demonstrated a very low (2.6%) rate of major adverse cardiac events (cardiac death, target vessel myocardial infarction, target lesion revascularisation) at nine-month follow-up, providing evidence of midterm safety and efficacy in 995 unselected real-world patients. In a substudy, using optical coherence tomography (OCT) analysis as a surrogate of vessel healing, the SUPRAFLEX stent showed 98% strut coverage at six-month follow-up6. Although thin-strut stents with a biodegradable polymer may play an important role in patients’ outcomes3, the SUPRAFLEX SES has never been tested in the context of a randomised outcome clinical trial. Also, for every technology seeking a share of the European market, a pre-specified set of steps must be undertaken in order to obtain the unconditional CE mark of approval7.
We aimed to compare clinical outcomes of the SUPRAFLEX with the current standard of care for atherosclerotic lesions (XIENCE drug-eluting stents; Abbott Vascular, Santa Clara, CA, USA) in broad patient and lesion scenarios from a “real-world” population.
Methods
STUDY OBJECTIVES
The TALENT trial (ClinicalTrials.gov: NCT02870140) was designed to assess the safety and effectiveness of the SUPRAFLEX sirolimus-eluting coronary stent system in a real-world consecutive all-comers patient population with symptomatic ischaemic heart disease undergoing PCI.
STUDY DESIGN
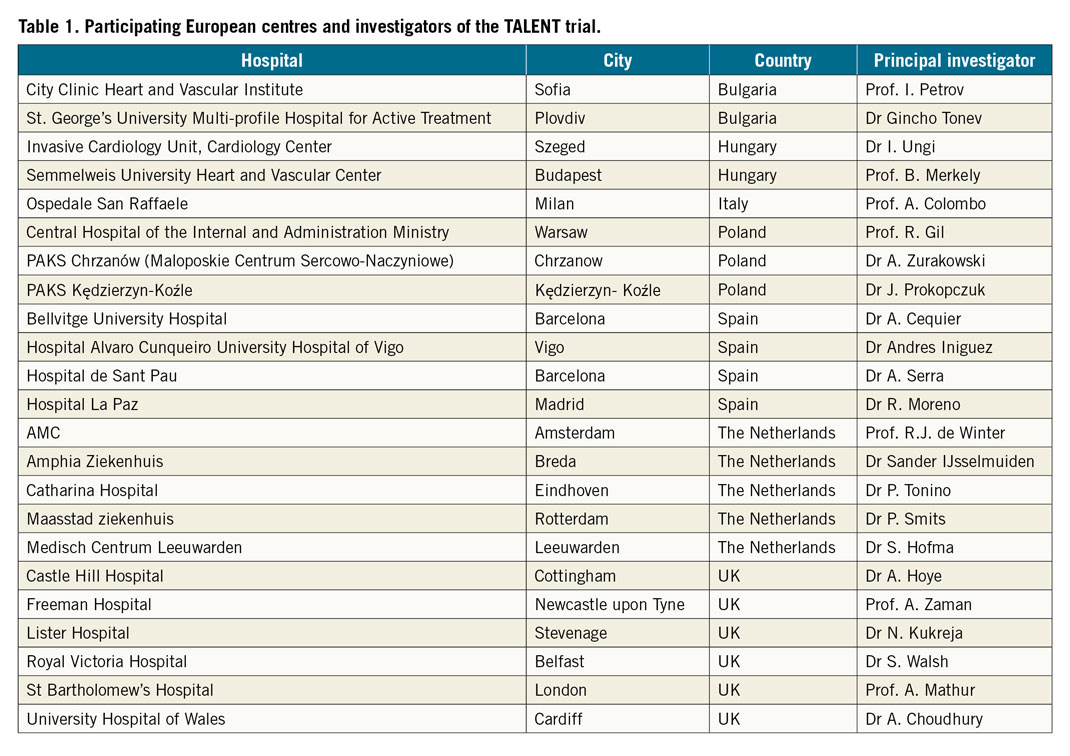
The TALENT trial is a prospective, single-blind (patient), randomised, 1:1 balanced, controlled, multicentre study. Twenty-three sites in Europe will participate (Table 1). The enrolment required for this study is 1,430 patients and, as there is a 1:1 randomisation, it is anticipated that 715 patients will be treated with the SUPRAFLEX and 715 patients will be treated with the control XIENCE family EES. Clinical data will be adjudicated by an independent clinical events committee. Randomisation will be performed via web-based software with random blocks according to centre. Randomisation will occur after all inclusion and exclusion criteria have been addressed and as soon as the baseline angiographic assessment confirms that the patient matches the enrolment criteria. All lesions for each patient treated at the index procedure should receive the same assigned stent type. The doctors responsible for the patients during their hospital stay were instructed to avoid telling the patients or writing in the discharge report to which treatment group the patients were assigned.
An independent Data Safety and Monitoring Board will monitor the individual and collective safety of the patients in the study during the enrolment phase and up to 12 months of follow-up (primary endpoint). The patients will be followed up to three years to assess their clinical status and major clinical events (Figure 1).

Figure 1. Flow chart of randomisation and follow-up in the TALENT trial.
PATIENT POPULATION
A total of 1,430 patients with symptomatic ischaemic heart disease who qualify for PCI are intended to participate in this study. Patients participating in the study must meet all of the following inclusion criteria: (i) patients must be 18 years of age or older, male or female; (ii) presence of one or more coronary artery stenoses of ≥50% in a native coronary artery or in a saphenous venous or arterial bypass conduit suitable for coronary stent implantation; (iii) the vessel should have a reference vessel diameter ranging from ≥2.25 mm to ≤4.5 mm, and (iv) the patients or legal guardian must understand all the trial requirements and provide written informed consent before any procedure is executed. Since this is an all-comers trial, inclusion criteria will be kept comprehensive to reflect routine clinical practice (“real-world, all-comer” patients); therefore, no restrictions are placed on the total number of treated lesions, treated vessels, lesion length, or number of stents implanted. Exclusion criteria are listed in Table 2.

STUDY ENDPOINTS
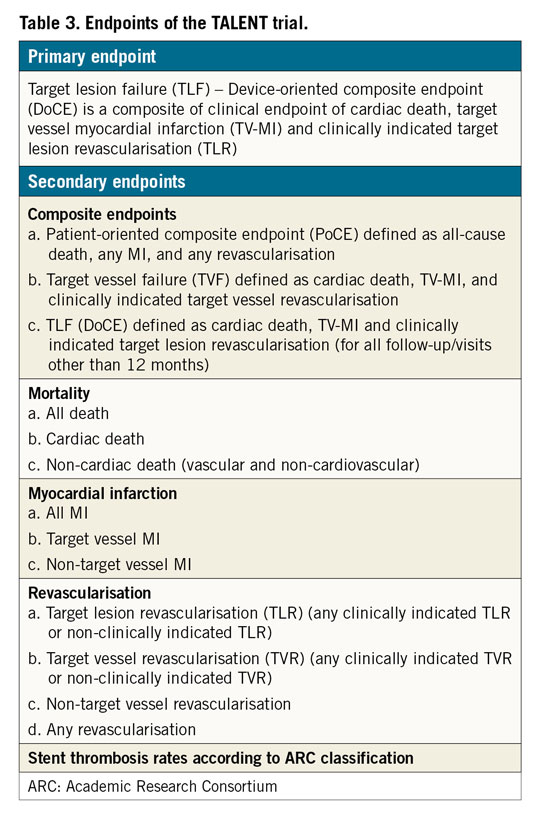
The primary endpoint for this trial is a non-inferiority comparison of the device-oriented composite endpoint target lesion failure (TLF) of the SUPRAFLEX group to the XIENCE group at 12 months post procedure. TLF (device-oriented composite endpoint [DoCE]) is a composite of (i) cardiac death, (ii) target vessel myocardial infarction (TV-MI), and (iii) clinically indicated target lesion revascularisation. Definitions of MI will follow the SCAI consensus for periprocedural MI (when ≤48 hours) or the third universal definition of MI >48 hours after the index procedure8,9. The secondary endpoints are described in Table 3.
DEVICES
The SUPRAFLEX is a CE-marked sirolimus-eluting coronary stent system with an L605 Co-Cr alloy coronary stent platform. The squared strut is 60 μm in thickness with highly flexible “S-link” connectors. The coating layer comprises the drug blended with a biodegradable polymeric matrix (poly L-lactide, 50/50 poly DL-lactide-co-glycolide and polyvinyl pyrrolidone). The average thickness of the coating ranges from 4 to 5 μm (Figure 2). The drug is 70% released within seven days and the remainder is released over a period of 48 days. The polymers gradually degrade over about nine to 12 months10 (Figure 3). This thin-strut biodegradable polymer stent will be tested against the XIENCE family of everolimus-eluting stents (e.g., XIENCE V®, XIENCE PRIME®, XIENCE Xpedition®, XIENCE PRO, XIENCE Alpine™ or any next generation of the XIENCE family; all Abbott Vascular). The control device is considered to be the current standard for DES given the ease of use and clinical performance, and also as preconised by the ESC executive summary on the evaluation of stents for post-marketing strategy before the unconditional CE mark7.
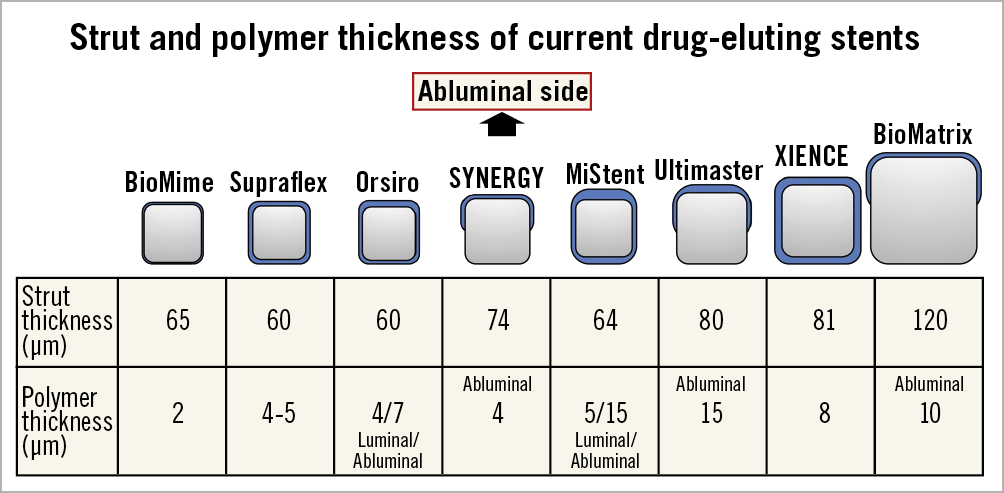
Figure 2. Schematic representation of strut and polymer thickness of contemporary stents.
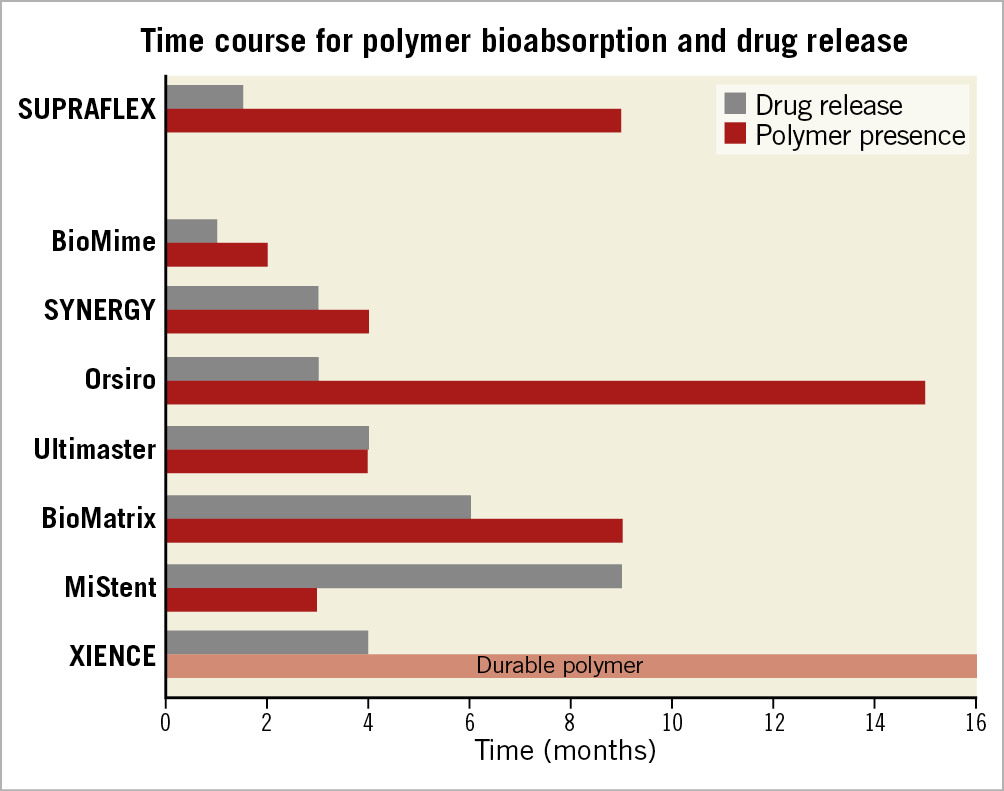
Figure 3. Bar graph showing the approximate time in months of drug release and presence of the polymer in the struts of contemporary drug-eluting stents with biodegradable polymers and the XIENCE stent.
INDEX AND STAGED PROCEDURES
The choice of size (length and diameter) of the stent will be left to the discretion of the interventionist and should cover the entire lesion. If multiple stents are required, the distal stent should be placed first and the second stent should be placed with a small (1-2 mm) overlap, avoiding gaps between them. In case of insufficient stent expansion, the stent will be post-dilated with an appropriately sized balloon. In case of an edge dissection, it is recommended to cover the dissection with the assigned study stent only. In case of a delivery failure, it is recommended first to try the comparator stent (crossover).
If the patient requires a planned staged procedure, this should be documented at the time of the index procedure (prior to the staged). The procedure must take place within 45 days after the index procedure for MI patients or 60 days for all other patients and must not be in the same epicardial vessel as any index lesion. For the staged procedure, the patient should receive the same stent as that assigned during the original index procedure (SUPRAFLEX or XIENCE)11.
PROCEDURE AND DEVICE SUCCESS
Acute device success (lesion basis) will be defined as a successful delivery and deployment of the assigned device at the intended target lesion and successful withdrawal of the delivery system with attainment of final in-stent residual stenosis of <30% (preferably by on-line QCA). Acute procedure success (patient basis) will be characterised by successful delivery and deployment of the assigned device at the intended target lesion and successful withdrawal of the delivery system with attainment of final in-stent residual stenosis of <30% (preferably by on-line QCA), without the occurrence of TLF during the index procedure hospital stay (maximum of seven days).
ADJUNCTIVE MEDICAL THERAPY
For patients eligible for PCI, a loading dose of 600 mg clopidogrel should be administered within 24 hours before the procedure. For those already under clopidogrel use for over five days, a loading dose of 300 mg may be administered at the physician’s discretion prior to the procedure. In accordance with the local standard of care, an alternative option to clopidogrel is the use of prasugrel or ticagrelor. In these cases, a loading dose of 60 mg of prasugrel or of 180 mg of ticagrelor at least two hours before the procedure can be used. Also, patients without chronic use of aspirin (ASA) must receive 100-300 mg (or dose per standard hospital practice) within 24 hours before the procedure12. For acute coronary syndromes (ACS), the order of preference is: (1) ticagrelor, (2) prasugrel (or clopidogrel) according to local practice and drug availability.
During the procedure, patients should receive unfractionated heparin (maintaining an activated clotting time of at least 250 sec during coronary angioplasty) or bivalirudin, according to the physician’s discretion and the site standard of care.
For post-procedural antiplatelet therapy, all stable coronary artery disease patients must receive dual antiplatelet therapy (DAPT), with ASA plus a P2Y12 inhibitor for at least six months after PCI followed by ASA monotherapy indefinitely. For those with ACS, DAPT should be given for at least 12 months after PCI followed by ASA monotherapy indefinitely. Extended DAPT will be at the discretion of the investigator.
STATISTICS
For the primary endpoint analysis, the intention-to-treat (ITT) population will be used, meaning that all patients will be analysed according to assigned treatment group, regardless of the treatment actually received. The ITT population will also be used for the primary analysis of all secondary clinical endpoints. A secondary analysis of the primary endpoint and all secondary clinical endpoints will also be conducted in the per protocol (PP) population. The PP population set will consist of all patients who have provided informed consent and have been randomised to a treatment group, and who have received only the stent assigned to that group. Subjects who do not receive a study stent, or who receive any stent other than the study stent to which they were randomised, will be excluded from the PP population.
The primary endpoint of TLF will be analysed and compared between the two treatment arms for non-inferiority of the SUPRAFLEX as compared to the control stent, XIENCE. The SUPRAFLEX expected TLF is based on the assumption of no difference in event rates between SUPRAFLEX and XIENCE. The study is powered at 85% to show non-inferiority for the SUPRAFLEX when compared to the XIENCE.
The 95% one-sided confidence interval for the difference in 12-month rates of TLF between the SUPRAFLEX and XIENCE arms will be calculated using Kaplan-Meier estimates at one year and their standard deviations. If this 95% interval excludes the non-inferiority margin, the SUPRAFLEX will be considered to be non-inferior to the XIENCE (this equates to a one-sided non-inferiority testing at alpha=5%).
The assumptions for the sample size calculation are as follows: a 1:1 treatment allocation ratio, a one-sided significance level (alpha) of 0.05, 85% power to show non-inferiority of SUPRAFLEX to XIENCE, a non-inferiority margin of 4%, a TLF event rate for XIENCE of 8.3% at 12 months13, no difference in event rate between the groups, and an attrition rate (loss to follow-up or withdrawal) of 3% (Figure 4).
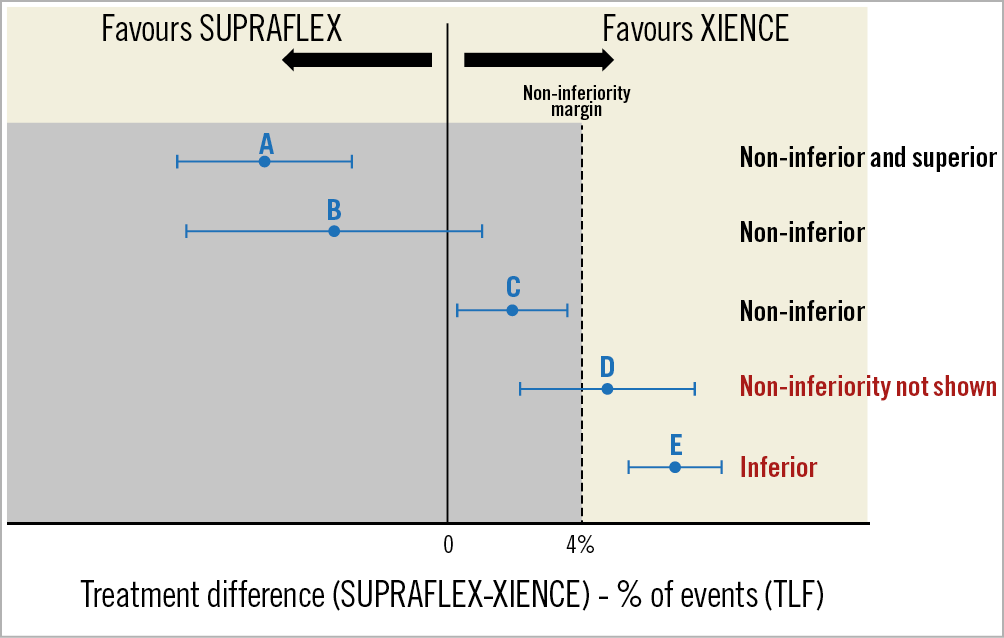
Figure 4. Representation of some possible results (examples A to E) of the non-inferiority analysis in the TALENT trial.
Using PASS software (NCSS LLC, Kaysville, UT, USA), the Farrington and Manning Likelihood Score Test requires 693 subjects in the SUPRAFLEX arm and 693 in the XIENCE arm, making a total of 1,386 patients. Taking into account an attrition rate –loss to follow-up– of approximately 3%, these numbers increase to 715 in each group, giving a total randomised sample of 1,430 patients.
Secondary endpoint analysis will occur at each follow-up visit and be compared between the SUPRAFLEX and XIENCE groups. For all clinical endpoints, the Kaplan-Meier method will be used. For all secondary endpoints conventional p-values and 95% confidence intervals for the difference in Kaplan-Meier results will be calculated. Dichotomous variables will be evaluated using Fisher’s exact test. Continuous variables will be evaluated by a two-sample t-test.
For time-dependent analyses, hazard ratios will be evaluated using a Cox proportional hazards model and Kaplan-Meier estimates will be evaluated according to the log-rank test.
For subgroup analysis of pre-specified subgroups, the SUPRAFLEX arm and the XIENCE arm will be compared on an ITT basis, dividing patients into the subgroups: diabetes, STEMI, small vessels (≤2.75 mm), multivessel treatment, long lesions (>18 mm), in-stent restenosis, bypass graft, left main treatment, bifurcation treatment, overlapping stents. For these subgroups, the primary endpoint will be evaluated. For these subgroups, the study does not have significant power to demonstrate non-inferiority for the SUPRAFLEX arm to the XIENCE arm, meaning that the results are considered exploratory (hypothesis-generating) only.
Discussion
For the evaluation of new coronary devices, the executive summary (task force on devices from the ESC) divides its requirements into pre- and post-market. The device used in this trial has been tested in the pre-market setting (first-in-man concept) and passed through an invasive imaging study6, necessary for the so-called conditional CE mark. For the post-market assessment, a randomised clinical outcome trial comparing the new device with the current standard of care must be performed, either with a superiority or a non-inferiority design. The results must be presented for the one-year primary clinical endpoint, with a mandatory continuous follow-up for five years. This is the final step for the unconditional CE-mark achievement7.
Also, a randomised clinical trial including a large sample is able to expose safety issues and peculiarities of the tested device related to technical issues. For instance, the recent DESSOLVE trial uncovered a dislodgement issue of the tested MiStent® (Micell Technologies, Durham, NC, USA)1.
These reasons are the pillars for performing this all-comers randomised clinical trial. The all-comers concept lies in the fact that no selection of patients (i.e., strict inclusion criteria) is performed. Thus, a “real-world” scenario can be resembled - in the case of coronary artery disease, no preference will emerge regarding stable disease or ACS, or regarding emergent, urgent or elective procedures. Although a new device (stent) all-comers trial theoretically mimics the real world, some issues are frequently encountered14, such as: (i) presence of a sufficient inventory of device sizes on the cath lab “shelf”; (ii) competitive trials - when a recruiting site participates in simultaneous stent trials, leading the enrolment in one of the trials to impact automatically on the recruitment for the other, provoking a specific type of selection bias; (iii) isolated investigator in a large cath lab team, meaning that a single interventionist recruits participants for that trial, whereas colleagues from the same site do not, leading to a deviation from the all-comers concept14.
For successful completion of a coronary device randomised clinical trial, precise evaluation of events such as MI and periprocedural myocardial infarction (PMI) are mandatory - especially since in clinical outcome trials they are almost unanimously part of the primary endpoint. The accurate detection of PMI demands careful interpretation of the definitions, because they can affect the primary outcome significantly. It has already been demonstrated that the number of events identified in clinical trials can be influenced by the specific definition of MI adopted. The proper collection of biomarkers (pre and post procedure), ECG data, and clinical status at baseline are crucial for the adequate identification of these events, thus achieving a credible event rate15.
As a sub-analysis of this study, an independent committee will assess retrospectively all the angiographies of reinterventions (target lesion revascularisation and target vessel revascularisation [TVR]), either clinically or non-clinically indicated revascularisation. For that, the investigators will use the off-line technique of quantitative flow ratio (qFR), a novel approach for assessing virtual fractional flow reserve (FFR) pullbacks from three-dimensional quantitative coronary angiography (QCA), precluding the use of a pressure wire16. This will enable a better understanding of the non-TVR events.
Limitations
Since this is a non-inferiority trial, assumptions for defining the margin had to be made. One of the assumptions was an event rate (primary endpoint - TLF) of 8.3%, derived from the RESOLUTE-AC trial13. However, it is important to bear in mind that, after the RESOLUTE-AC trial, evolution in stent technology, intraprocedural imaging techniques and adjunctive medical therapy (e.g., novel antiplatelet therapy) might have been responsible for reducing this rate. Thus, it should be no surprise to encounter a lower event rate in TALENT, similar to the rate found in the DESSOLVE III trial1 of 6.5% in the control group, with a potential bias compared to the previous choice of the non-inferiority margin – 50% of the event rate17. In that case, the p-value for a lower non-inferiority margin can be calculated (e.g., 50% of the overall event rate in the trial) as a sensitivity analysis.
Conclusions
The trials concerning the new-generation DES have demonstrated that these new devices have already reached a plateau of outcomes, with low rates of stent thrombosis (<1%) and target lesion revascularisation (<5%). Thus, demonstrating superiority over the standard of care becomes challenging, and a non-inferiority trial is the only viable statistical tool. Some unmet specific and surrogate goals are still to be unravelled, e.g., lack of strut coverage. Since this phenomenon relates to stent thrombosis18, and previous OCT reports have demonstrated that the SUPRAFLEX struts are 98.1% covered after six months, outshining rates for the XIENCE (94.1%) and PROMUS Element™ (91.5%) (Boston Scientific, Marlborough, MA, USA), the “surrogate outcome” of these OCT findings could potentially emerge in the long-term clinical outcome6.
|
Impact on daily practice The TALENT trial mimics the real world of patients undergoing percutaneous coronary intervention with DES – with an all-comers principle. The intention of the trial is to show non-inferiority of a thinner strut with biodegradable polymer (SUPRAFLEX) compared to its standard-of-care counterpart, XIENCE. Demonstrating the safety and effectiveness of this device could be groundbreaking for a better understanding of and for advancing studies with this technology. |
Guest Editor
This paper was guest edited by Alec Vahanian, MD, PhD; Department of Cardiology, Hôpital Bichat-Claude Bernard and University Paris VII, Paris, France.
Funding
TALENT is an investigator-initiated trial sponsored by the European Clinical Research Institute (www.ECRI-trials.com), which received funding from SMT (Sahajanand Medical Technologies Pvt. Ltd., India).
Conflict of interest statement
P.W. Serruys reports personal fees from Abbott Laboratories. Y. Onuma is a member of the Advisory Board of Abbott Vascular. The other authors have no conflicts of interest to declare. The Guest Editor is a consultant for Edwards Lifesciences.
