
Abstract
Aims: The aim of this study was to evaluate the safety and efficacy of the BioMime sirolimus-eluting coronary stent (SES) compared to the XIENCE family of everolimus-eluting coronary stents (EES) in the treatment of patients with de novo native coronary artery lesions.
Methods and results: The meriT-V is a prospective, multicentre, randomised, open-label, active-controlled, non-inferiority trial. A total of 256 patients with up to two de novo native coronary artery lesions were enrolled and randomly assigned (2:1) to BioMime SES or XIENCE EES. BioMime SES was non-inferior to XIENCE EES for the primary endpoint of in-stent late lumen loss (0.15±0.27 mm vs. 0.15±0.29 mm; difference: -0.006 mm; 95% confidence interval: -0.085 to 0.072; p=0.87; p for non-inferiority <0.0001) at nine-month follow-up. The major adverse cardiac events rate was numerically lower in the BioMime SES group (2.98% vs. 7.14%; p=0.13), driven by a statistically significant lower risk of any myocardial infarction (0.60% vs. 4.76%; p=0.03), when compared with the XIENCE EES group. There was no difference in target vessel myocardial infarction (p=0.62) between the groups. There was no definite or probable stent thrombosis in either group.
Conclusions: In the treatment of de novo native coronary artery lesions, the biodegradable polymer ultra-thin SES (BioMime) was non-inferior to a durable polymer EES (XIENCE) at nine-month follow-up. Further studies powered for clinical endpoints are needed.
Abbreviations
CFD: cumulative frequency distribution
DES: drug-eluting stent(s)
EES: everolimus-eluting coronary stent(s)
ID-TLR: ischaemia-driven target lesion revascularisation
ID-TVR: ischaemia-driven target vessel revascularisation
LLL: late lumen loss
MACE: major adverse cardiac events
MLD: minimum lumen diameter
PCI: percutaneous coronary interventions
QCA: quantitative coronary angiography
SES: sirolimus-eluting coronary stent
Introduction
Drug-eluting stents (DES) represent a key advance in percutaneous coronary interventions (PCI) owing to their ability to inhibit neointimal proliferation, which lowers the need for repeat revascularisation1,2. However, older-generation DES have been shown to increase the risk of late restenosis and stent thrombosis. Efforts to reduce these risks include improvements in stent platforms, polymer carriers, and drug selection. Thinner struts reduce vessel wall injury, decrease inflammation and promote faster endothelialisation.
The second-generation thin-strut DES have been shown to reduce the risk of restenosis, stent thrombosis and myocardial infarction (MI) or possibly death when compared with older-generation DES or bare metal stents3,4. Moreover, the newer generation of biodegradable polymer stents has the potential to reduce the inflammatory reaction of the arterial wall and minimise the risk of late restenosis and thrombus formation5,6. More recently, ultra-thin (<70 µm) DES have been shown to improve outcomes further compared with second-generation DES7.
The BioMime™ (Meril Life Sciences Pvt. Ltd., Vapi, India) is an ultra-thin sirolimus-eluting coronary stent (SES) with an established preliminary safety and efficacy record in the previous meriT-1, meriT-2 and meriT-3 trials in treating single de novo and complex lesions8-10. In the present randomised study, we compared the safety and efficacy of the biodegradable polymer-coated BioMime SES with the XIENCE family (XIENCE V®, PRIME® or Xpedition®; Abbott Vascular, Santa Clara, CA, USA) of durable polymer-coated everolimus-eluting coronary stents (EES) in patients undergoing PCI with up to two de novo native coronary artery lesions.
Methods
DEVICE DESCRIPTION
The BioMime is an ultra-thin strut (65 µm) SES that uses a cobalt-chromium platform with a unique hybrid design of open cells in the mid segment and closed cells at the edges, coated with biocompatible and bioabsorbable polymers, PLLA (poly-L-lactic acid) and PLGA (poly-lactic-co-glycolic acid). Overall, this polymer formulation provides a uniformly thin coating (2 µm) which degrades in approximately six to nine months after implantation. Moreover, despite thinner struts, the radial strength (1,320 mmHg for size 3.00x44 mm) of the BioMime is preserved with minimal stent recoil (<3%) and foreshortening (0.29%) due to the unique hybrid cell design of open and closed cells. The antiproliferative drug, sirolimus, is coated on the stent surface area at a dose of 1.25 μg/mm2 which is released within 30-40 days after stent implantation.
STUDY DESIGN AND PATIENT POPULATION
The meriT-V is a prospective, multicentre, randomised, open-label, active-controlled, non-inferiority trial (ClinicalTrials.gov Identifier: NCT02112981), which enrolled 256 patients from 15 investigational sites in Europe (12 sites, including the Netherlands, Belgium, UK, Spain, Latvia, FYR of Macedonia, Czech Republic, and Poland) and Brazil (three sites) between November 2014 and December 2016. The enrolled patients were randomised (2:1) to BioMime SES or XIENCE EES (XIENCE V/PRIME/Xpedition). The unequal randomisation provides a larger sample size in the BioMime SES group in order to evaluate adverse effects and allow a more precise estimation of any learning effects with this new device. The study was conducted in accordance with the Declaration of Helsinki, Good Clinical Practice (E6), and ISO14155. The clinical trial protocol was approved by the institutional review board at each investigational site and all patients provided written informed consent before inclusion in the study. Clinical follow-up was scheduled at 30 days, 5 months, 9 months, 12 months and 24 months post procedure. Angiographic follow-up of all patients was scheduled at nine months post procedure. The data obtained were managed and analysed by an independent contract research organisation (JSS Medical Research India Pvt. Ltd., Faridabad, India).
The eligibility criteria included age ≥18 years and presence of ischaemic heart disease or myocardial ischaemia (stable angina, unstable angina, or silent ischaemia) with up to two de novo native coronary artery lesions (length ≤44 mm) and a reference vessel diameter ≥2.5 and ≤3.5 mm. The key exclusion criteria were left main or aorto-ostial location; left ventricular ejection fraction ≤30%; extreme vessel tortuosity or lesion angulation (˂45˚); severe calcification proximal to or within the target lesion; restenotic lesion and bifurcation lesions with side branch diameter >2 mm. In addition, patients with evidence of an acute Q-wave or non-Q-wave MI within 72 hours preceding the index procedure were excluded, unless the CK and CK-MB enzymes were less than twice the upper limit of normal.
RANDOMISATION AND PROCEDURES
All patients were randomised in a 2:1 ratio to either the BioMime SES or the XIENCE EES using a central web-based data capture system (block of 6). Before catheterisation, patients were administered aspirin (75 mg) and a loading dose of clopidogrel (600 mg). Heparin was administered to maintain an activated clotting time of ~250-300 seconds or 150-200 seconds if a glycoprotein IIb/IIIa inhibitor was administered. The BioMime SES was available in lengths of 8-48 mm and diameters of 2.00-4.50 mm; the XIENCE EES was available in lengths of 8-38 mm and diameters of 2.25-4.00 mm. Post-dilatation was mandatory and performed within the deployed stent to optimise stent expansion. Dual antiplatelet therapy (DAPT) consisting of aspirin (75-150 mg/day) and clopidogrel (75 mg/day) or prasugrel (10 mg/day) or ticagrelor (90 mg/day) was prescribed for a minimum duration of one year. Beyond one year, aspirin was recommended for an indefinite period.
STUDY ENDPOINTS AND DEFINITIONS
The primary angiographic endpoint was in-stent late lumen loss (LLL) at nine months. The secondary angiographic endpoints were in-stent and in-segment diameter stenosis, minimum lumen diameter (MLD) and in-segment LLL at nine months. Late lumen loss was defined as the difference between the MLD after stent implantation and at nine months post procedure. Binary restenosis was defined as >50% diameter stenosis. The clinical endpoint was major adverse cardiac events (MACE), defined as a composite of cardiac death, any MI and ischaemia-driven target vessel revascularisation (ID-TVR). Cardiac death was defined as any death due to immediate MI, arrhythmia, cardiac failure, cardiac arrest, deaths related to the procedure, or unknown cause. MI was defined as symptoms indicative of ischaemia/infarction in association with electrocardiogram (ECG), cardiac biomarker or pathologic evidence of infarction. Periprocedural MI was defined as total creatine kinase muscle/brain (CK-MB) elevation >3× the upper reference limit. ID-TVR was defined as repeat PCI or coronary artery bypass grafting of the target vessel (TV) associated with ≥50% diameter reduction together with documented ischaemia. Ischaemia-driven target lesion revascularisation (ID-TLR) was defined as revascularisation associated with a diameter stenosis (DS) of ≥50% with ischaemia or symptoms, or a diameter stenosis of ≥70% observed even without any signs and symptoms of ischaemia at the time of follow-up angiography. Stent thrombosis was identified and categorised according to the definitions provided by the Academic Research Consortium11. Procedural success was defined as angiographic success along with absence of MACE during index hospitalisation. All events related to endpoints were confirmed by an independent adjudication committee.
QUANTITATIVE CORONARY ANGIOGRAPHY
Coronary angiograms recorded at baseline, post procedure, and at nine-month follow-up were analysed by an independent angiographic core laboratory (Cardiovascular Research Centre, Sao Paulo, Brazil) using an automated edge detection system (Medis medical imaging systems, Leiden, the Netherlands) with standard methodology for DES analysis, as previously reported12. In each target lesion, paired QCA was performed within the stent and the analysis segment (including the stented region and 5 mm edge regions) and reported separately. The core lab was blinded to the randomised stent assignment.
STATISTICAL ANALYSIS
The meriT-V randomised study was powered for non-inferiority of the BioMime SES compared with the XIENCE EES for the primary endpoint of in-stent LLL at nine months. Assuming a dropout rate of 10%, a total of 256 patients were randomly assigned 2:1 to treatment with BioMime SES (170 patients) or XIENCE EES (86 patients) that provided 90% power at a one-sided alpha (α) of 0.05 with a non-inferiority margin of 0.195 mm. The non-inferiority margin was considered to yield less than one half of the estimated standard deviation of in-stent LLL (0.41 mm) for the XIENCE arm, as estimated in the SPIRIT III trial13. A post hoc power calculation based on actual dropout rates for angiographic follow-up showed that the trial had 86% power to demonstrate non-inferiority of the BioMime SES when compared with the XIENCE EES. Results were reported as mean±standard deviation for continuous variables and number (%) for categorical variables. Continuous variables with a normal distribution were compared using the Student’s t-test for independent samples and, if the distribution was not normal, the Mann-Whitney U test was used. Categorical variables were compared using the chi-square test or Fisher’s exact test. MACE-free survival rates were calculated using the Kaplan-Meier method and compared using the log-rank test. A p-value <0.05 was considered statistically significant.
Results
A total of 256 patients were enrolled and randomly assigned (2:1) to the BioMime SES group (170 patients, 182 lesions) or the XIENCE EES group (86 patients, 95 lesions). Baseline clinical, procedural and lesion characteristics were comparable between the treatment groups (Table 1, Table 2). Mean age was 64.33±9.57 and 64.70±8.99 years in the BioMime SES and XIENCE EES groups, respectively. There was a slightly higher prevalence of diabetes mellitus in the BioMime SES group (24.12%; n=41) as compared to the XIENCE EES group (20.93%; n=18).
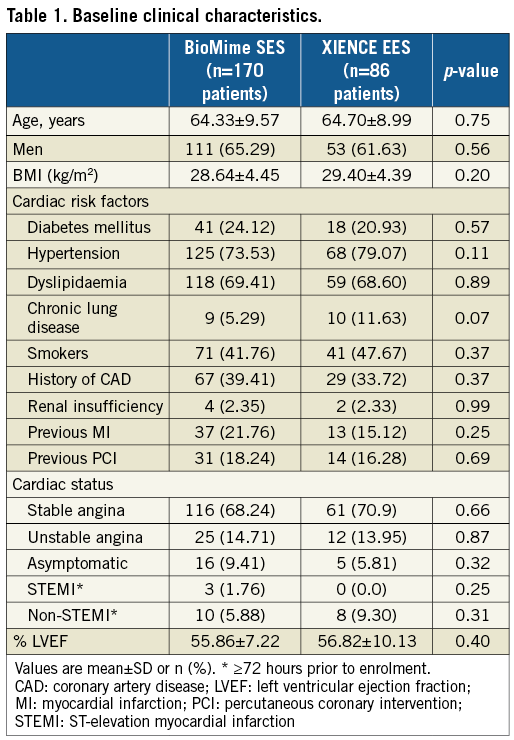
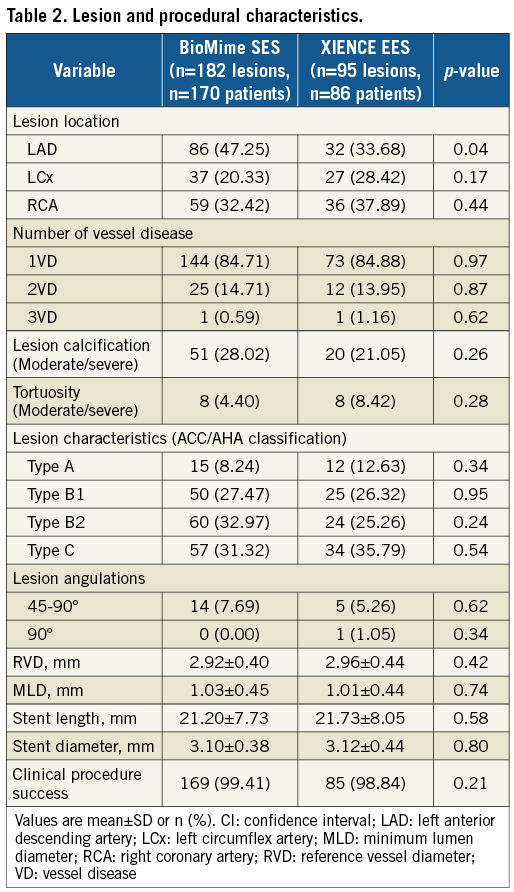
QCA results at baseline, post procedure, and nine-month angiographic follow-up are summarised in Table 3. Angiographic follow-up at nine months was completed in 201 (78.52%) patients, of whom 131 patients (146 lesions) and 70 patients (79 lesions) were included in the BioMime SES group and the XIENCE EES group, respectively (Figure 1). However, data for two patients (three lesions) in the XIENCE EES group and one lesion (out of two lesions in one patient) in the BioMime SES group were not analysable. As a result, the primary endpoint of in-stent LLL was measured in 199 patients (BioMime SES: 131 patients, 145 lesions; XIENCE EES: 68 patients, 76 lesions). In-stent LLL was 0.15±0.27 mm in the BioMime SES group and 0.15±0.29 mm in the XIENCE EES group, which demonstrates non-inferiority of the BioMime SES compared to the XIENCE EES (difference: −0.006 mm; 95% confidence interval: −0.085 to 0.072; p=0.87: p-value for non-inferiority <0.0001). Moreover, the secondary endpoints of in-stent and in-segment MLD and %DS were non-significantly different between the BioMime SES and XIENCE EES groups (Table 3). The cumulative frequency distribution (CFD) curve of in-stent LLL up to nine months is illustrated in Figure 2. Of note, of the four patients with ID-TLR in the BioMime SES group, in-stent LLL data were available for only two patients as the other two patients withdrew consent.
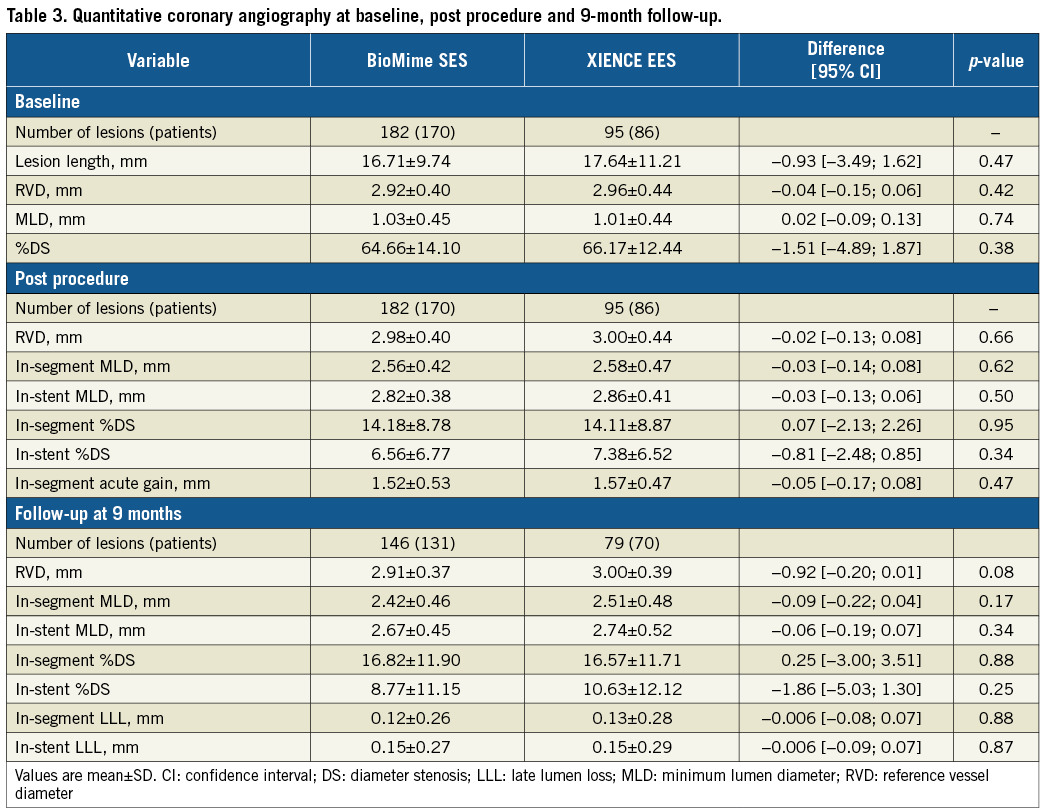
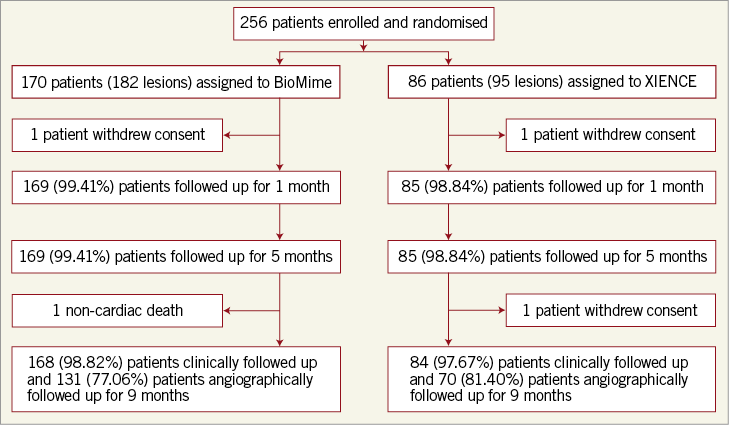
Figure 1. Patient enrolment and disposition.
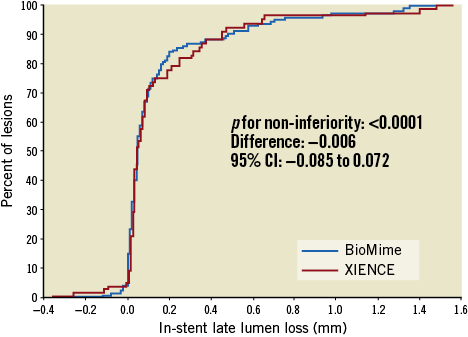
Figure 2. Cumulative frequency distribution curves for in-stent late lumen loss.
Overall, the device success rate was 99.41%, because in one patient the BioMime SES was unable to cross the lesion; hence, the patient was implanted with another DES. Clinical follow-up at nine months was available for 252 (98.44%) patients (168 patients in the BioMime SES arm and 84 patients in the XIENCE EES arm). The MACE rate was non-significantly lower in the BioMime SES group (2.98% vs. 7.14%; p=0.13). Moreover, there was a significantly lower risk of any MI (0.60% vs. 4.76%; p=0.03) in the BioMime SES group when compared with the XIENCE EES group (Table 4, Figure 3). However, the incidence of target vessel myocardial infarction (TV-MI) (0.60% vs. 1.19%; p=0.62) was not different between the two groups.
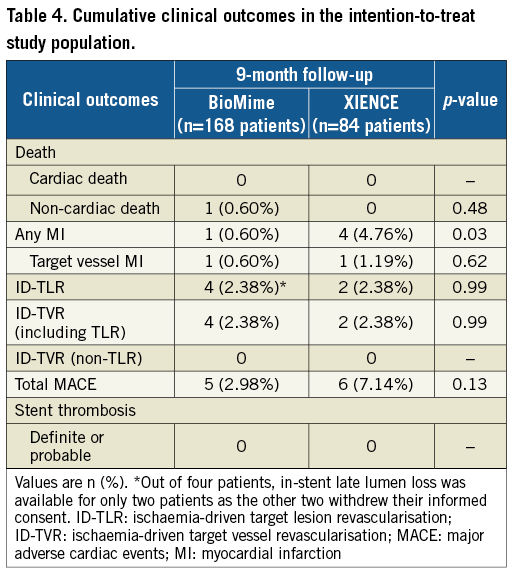
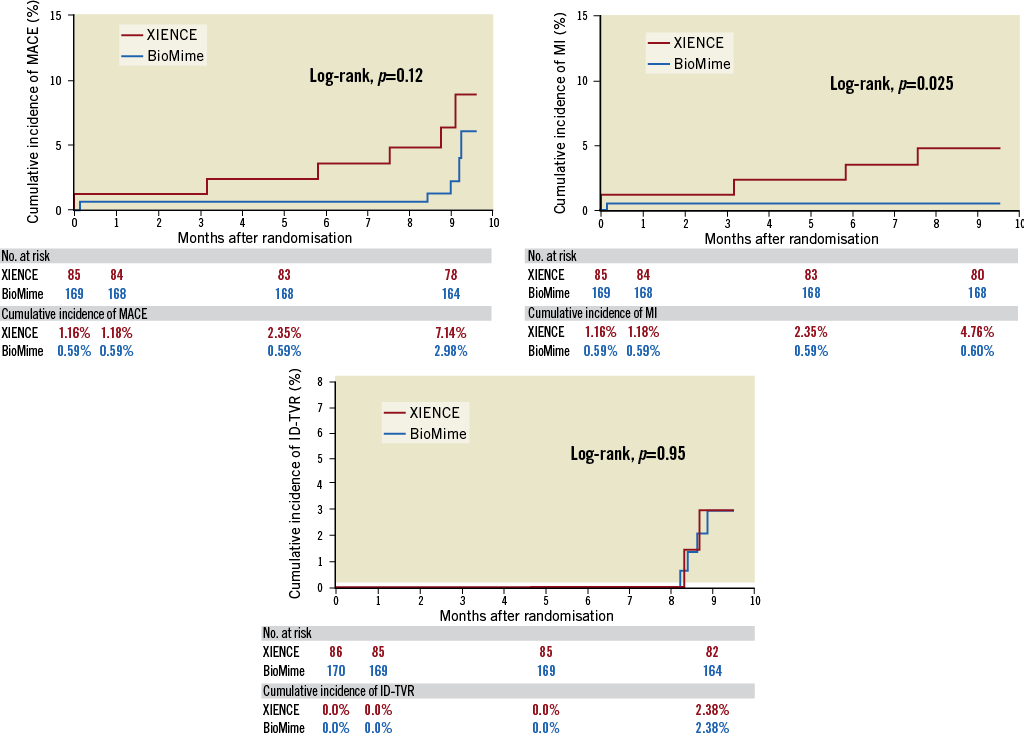
Figure 3. Kaplan-Meier cumulative event curves of major adverse cardiac events (MACE), myocardial infarction (MI) and ischaemia-driven target vessel revascularisation (ID-TVR) for the sirolimus-eluting coronary stent (SES) vs. the everolimus-eluting coronary stent (EES).
There was one non-cardiac death due to cerebral stroke in the BioMime SES group; no death was reported in the XIENCE EES group at nine months post procedure. In the BioMime SES group, one patient presented with a periprocedural MI. The same patient experienced ID-TLR in the form of PCI at nine months post procedure. There was no definite or probable stent thrombosis in either of the two treatment groups. There was no stent fracture on visual angiographic assessment with either of the stent platforms at follow-up.
Discussion
In the meriT-V randomised trial, the BioMime SES was non-inferior to the XIENCE EES for the primary angiographic endpoint of in-stent LLL. Moreover, the findings from the secondary angiographic endpoints (%DS, MLD, and in-segment LLL) demonstrated that the BioMime SES was comparable with the XIENCE EES. Safety outcomes, including the rates of MACE, cardiac death, ID-TVR, and stent thrombosis, occurred with similar frequency in the BioMime SES and XIENCE EES groups. There was a notably lower rate of any MI in the BioMime as compared with the XIENCE without any difference in TV-MI. Of note, the trial was not powered for clinical endpoints and the results are hypothesis-generating.
Although first-generation DES significantly reduced restenosis when compared with bare metal stents, the late catch-up phenomenon, late restenosis, and stent thrombosis were observed. Causal mechanisms for the increased risk included thicker struts and less biocompatible polymers leading to inflammatory reactions. Given this limitation, biodegradable polymer DES were developed. The basis of these devices was to provide early efficacy similar to that of a durable polymer DES, with the additional advantage of late safety after degradation of the polymer. The safety and performance of the BioMime SES have been evaluated in three previous clinical trials, namely meriT-1, meriT-2, and meriT-3. In the meriT-1 trial, in which 30 patients were enrolled, the study showed the absence of MACE and stent thrombosis at one-year follow-up. Median in-stent LLL (primary endpoint) was 0.15 mm at eight months with no case of binary restenosis8. In the meriT-2 trial, 250 patients were enrolled and had PCI using the BioMime SES stent. At eight-month angiographic follow-up, median in-stent LLL was 0.12 mm. At one-year follow-up, clinically indicated TLR was reported in 12 (4.8%) patients9. In the meriT-3 trial, in which 1,161 all-comer patients were enrolled, the study showed 26 (2.35%) incidences of MACE and only one (0.09%) case of stent thrombosis at one-year follow-up10.
The meriT-V trial was designed to compare the biodegradable polymer SES (BioMime) against the durable polymer EES (XIENCE) for the angiographic endpoint of in-stent LLL at nine months. The trial showed that the BioMime SES was non-inferior to the XIENCE EES for the primary endpoint of in-stent LLL (p for non-inferiority <0.0001). Moreover, ID-TLR, which could be regarded as the most appropriate efficacy measure of DES, was low and similar between BioMime (2.38%) and XIENCE (2.38%) at nine months post procedure. Likewise, in the DESSOLVE III trial, SES vs. EES demonstrated a similar rate of TLR (2.14% vs. 3.45%; p=0.16) at one year after PCI14. An angiographic study in the ORIENT trial (randomised evaluation of the Orsiro SES vs. the Resolute Integrity zotarolimus-eluting stent [ZES]) enrolling 372 patients demonstrated comparable results for median in-stent LLL (0.06 mm [−0.09 to 0.24] vs. 0.12 mm [−0.07 to 0.32 mm]; p=0.205) at nine months after the index procedure15. In the BIOFLOW-II trial, the Orsiro SES (Biotronik AG, Bülach, Switzerland) was non-inferior to the XIENCE EES for in-stent LLL at nine months (0.10±0.32 mm vs. 0.11±0.29 mm; difference: 0.00063 mm; 95% confidence interval: −0.06 to 0.07; p=0.98; p for non-inferiority <0.0001)16. Overall, the results of meriT-V are consistent with those observed in previous randomised studies between bioresorbable polymer SES and durable polymer EES.
Our study had a relatively low incidence of MACE in both treatment groups. There was a good adherence to DAPT in both the BioMime and XIENCE groups (99.41% vs. 100%, respectively). Of note, the rate of MACE was numerically lower with BioMime SES when compared with XIENCE EES, driven by a statistically significant reduction in MI (p=0.03) without any difference in TV-MI. Other studies, such as the BIOFLOW V trial, showed superiority of the Orsiro stent (61 µm) to the XIENCE stent (81 µm) with a reduction in TV failure driven by a decrease in MI17. A recent meta-analysis of ten randomised trials comparing ultra-thin DES (Orsiro, MiStent [STENTYS, Paris, France] and BioMime) vs. thicker second-generation DES (XIENCE, Resolute Integrity® [Medtronic, Minneapolis, MN, USA] and Nobori® [Terumo Corp., Tokyo, Japan]) showed reduced target lesion failure at one year driven by less MI and numerically lower stent thrombosis with the ultra-thin DES, raising the hypothesis that further reduction in strut thickness might be beneficial7. Although the precise mechanism of reduced MI in these studies is not clearly established, thinner struts may result in less MI7.
Study limitations
The study was powered for the primary angiographic endpoint and not for clinical endpoints. Moreover, the dropout rate for angiographic follow-up was more than the 10% projected during the initial sample size calculation. Such a phenomenon reflects the increasing difficulty to have patients returning for invasive procedures without an objective clinical need, despite being on a clinical study. However, a post hoc power calculation using actual dropout rates showed that the trial was still adequately powered to establish non-inferiority of the BioMime SES vs. the XIENCE EES for the primary endpoint of in-stent LLL. The TLR rate was probably driven by angiographic follow-up which may be the result of oculostenotic reflex. However, the trial was not designed or powered for clinical endpoints. In addition, there was no difference in LLL between the two stent groups and hence this “oculostenotic” reflex should not differentially affect the rate of TLR between the two arms.
Conclusions
In patients with coronary artery disease, the biodegradable polymer ultra-thin SES (BioMime) was non-inferior to the durable polymer EES (XIENCE) for the angiographic endpoint of in-stent LLL. The BioMime SES group had a lower MACE rate, along with a significantly lower rate of any MI (but no difference in TV-MI), than the XIENCE EES group. There was no definite or probable stent thrombosis in either stent group. These results provide the basis for further randomised trials to test whether the BioMime SES could be superior to the current generation of durable polymer DES for clinical outcomes.
| Impact on daily practice Compared with the durable polymer EES (XIENCE V, PRIME or Xpedition) of the XIENCE family, the biodegradable polymer ultra-thin SES (BioMime) elucidated non-inferiority based on the angiographic endpoint of in-stent late lumen loss. The BioMime SES group demonstrated a lower rate of MACE while any MI was significantly lower than that of the XIENCE EES group. |
Acknowledgements
We thank Drs A. IJsselmuiden, J. Koolen, P. Kala and L. Janssens who were invaluable collaborators in the execution of the meriT-V trial.
Funding
The meriT-V trial was funded by Meril Life Sciences Pvt. Ltd., India.
Conflict of interest statement
A. Abizaid and R. Costa are external scientific advisors to Meril Life Sciences Pvt. Ltd., Vapi, India. A. Abizaid is also the principal investigator for the meriT-V trial. S. Bangalore is on the advisory board of and has received research grants and honoraria from Abbott Vascular. The other authors have no conflicts of interest to declare.
