Abstract
Aims: The study sought to evaluate the safety and efficacy of FIREHAWK, a novel abluminal groove-filled biodegradable polymer sirolimus-eluting stent (SES) for treating patients with single de novo coronary lesions compared with the durable polymer everolimus-eluting stent (EES) XIENCE V.
Methods and results: A total of 458 patients with single de novo native coronary lesions ≤24 mm in length and a coronary artery ≥2.25 to ≤4.0 mm in diameter were enrolled in the TARGET I study, a prospective, randomised, non-inferiority trial. The primary endpoint was in-stent late lumen loss (LLL) at nine-month follow-up. The secondary endpoint, target lesion failure (TLF), was defined as the composite of cardiac death, target vessel myocardial infarction (TVMI), or ischaemia-driven target lesion revascularisation (iTLR). Patients were centrally randomised to treatment with either biodegradable polymer SES (n=227) or durable polymer EES (n=231). The nine-month in-stent LLL of the biodegradable polymer SES was comparable to the EES group (0.13±0.24 mm vs. 0.13±0.18 mm, p=0.94; difference and 95% confidence interval 0.00 [-0.04, 0.04] mm; p for non-inferiority <0.0001). Cardiac death (0.4% vs. 0.0%), TVMI (1.3% vs. 1.7%), iTLR (0.4% vs. 0.4%) and TLF (2.2% vs. 2.2%) were similar between the biodegradable polymer SES and durable polymer EES groups at 12-month follow-up (all p>0.05). No definite/probable stent thrombosis was observed in both of these groups.
Conclusions: In the multicentre TARGET I trial, the novel abluminal groove-filled biodegradable polymer SES FIREHAWK was non-inferior to the durable polymer EES XIENCE V with respect to the primary endpoint of in-stent LLL at nine months for treating patients with single de novo coronary lesions. The incidences of clinical endpoints were low in both of the stents at 12-month follow-up. (ClinicalTrials.gov identifier: NCT01196819)
Introduction
Drug-eluting stents (DES) delivering antiproliferative drugs from durable polymer have significantly reduced angiographic and clinical measures of restenosis compared with bare metal stents, with the low risk of adverse events including myocardial infarction (MI) and death1-5. However, durable polymers of first-generation DES have been linked to persistent inflammation, and delayed endothelial healing may result in an increased risk of late and very late stent thrombosis (ST)6. Recent advances in stent technology and use of invasive imaging techniques, along with the introduction of biocompatible or biodegradable polymers of newer-generation DES, have minimised the risk of complications compared with the first-generation DES7-12.
The FIREHAWK® (MicroPort Medical, Shanghai, China) biodegradable polymer sirolimus-eluting stent (SES) is a novel abluminal groove-filled biodegradable poly-lactic acid (PLA) polymer DES with specific targeting of sirolimus release13,14. The TARGET I study is the first randomised trial that aimed to assess the safety and efficacy of the FIREHAWK biodegradable polymer SES compared with the durable polymer everolimus-eluting stent (EES) XIENCE V® (Abbott Vascular, Santa Clara, CA, USA) in treatment of patients with single de novo coronary lesions.
Methods
STUDY DESIGN AND PATIENT SELECTION
The TARGET I study is a prospective, multicentre, randomised trial, which enrolled 458 patients with single de novo coronary artery lesions in a 1:1 ratio to receive either the FIREHAWK stent or the XIENCE V stent at 16 high-volume medical centres in China. Patients were eligible for enrolment if they were 18 years or older and intended to undergo PCI treatment with a single de novo native coronary artery lesion. The target lesion had a diameter stenosis ≥70%, a reference vessel diameter between 2.5 and 4.0 mm, and lesion length ≤24 mm by visual estimation. The major exclusion criteria included acute myocardial infarction within one week, chronic total occlusion, left main coronary artery, bifurcation (side branch ≥2.5 mm in diameter), in-stent restenosis. Clinical follow-up was planned to be performed at 30 days, six months, nine months, one year and then annually up to five years after the index procedure. Angiographic follow-up was conducted at nine months ±30 days post-index procedure. The trial was approved by the institutional ethics committee at each participating site. All eligible patients signed written informed consent for participation in the trial. The trial is registered with ClinicalTrials.gov identifier: NCT01196819.
STUDY DEVICES
The FIREHAWK stent (MicroPort Medical, Shanghai, China) is pre-mounted on a rapid-exchange delivery system, and consists of three components: a cobalt-chromium L605 platform, PLA polymer, and the antiproliferative drug sirolimus. Details of the design features have been previously described13. Briefly, the unique abluminal grooves are scored at the outer surface of the struts (total strut thickness: 86 μm), with average sirolimus dosage of 3 µg/mm stent lengths (Figure 1). The preclinical study showed that the drug release kinetics curves were similar between the FIREHAWK and CYPHER stents (Online Figure 1). The FIREHAWK stent was available in diameters of 2.25, 2.5, 2.75, 3.0, 3.5 and 4.0 mm and in lengths of 13, 18, 23 and 29 mm. The XIENCE V stent is comprised of a medical grade L-605 cobalt-chromium alloy backbone and delivery system, a thin fluoropolymer coating and everolimus (100 µg of everolimus per square centimetre of stent surface area). The total strut thickness of the XIENCE V is 89 µm, which consists of metal thickness of 81 µm and polymer thickness of 8 µm. The XIENCE V stent was available in diameters of 2.25, 2.5, 2.75, 3.0, 3.5 and 4.0 mm and in lengths of 12, 15, 18, 23 and 28 mm.

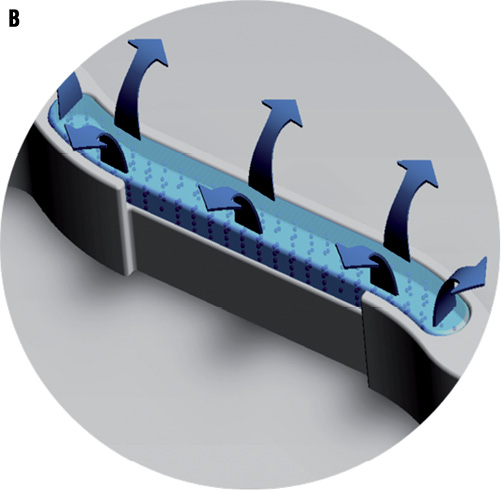
Figure 1. A micrograph and illustration of the FIREHAWK stent. A) Micrograph: FIREHAWK stents employ an “S” type design in the connecting rods, which simultaneously makes the stent sufficiently flexible, reducing the distance between the sine curve circles, increasing the percentage of the metal coverage of the stent and allowing for coverage of the lesion (scanning electron microscopy, original magnification ×150). B) This illustration of the FIREHAWK stent shows the slotted, one-sided, coated design. The slots play a fixed role concerning the biodegradable coating and the loaded drug on the vessel wall side can be target released.
RANDOMISATION, PROCEDURES AND ADJUNCT DRUG THERAPY
Patients who met the inclusion and exclusion criteria were stratified by centre using the fixed block method and were randomised 1:1 using a web-based randomisation system to treatment with the FIREHAWK biodegradable polymer SES or the XIENCE V durable polymer EES. The clinical events committee and independent angiographic core laboratory, but not the investigator or the patient, were blinded to the assigned study stent.
Lesions were treated by standard interventional techniques. Pre-dilation and post-dilation were left to the discretion of the investigator. In the event of a bail-out procedure and additional stent requirement, the stent had to be one from the same group as the first implanted stent.
Prior to stent implantation, all patients received treatment with aspirin (300 mg, at least 24 hours before the intervention) and clopidogrel (loading dose: 300 mg, at least six hours before the intervention; for those having taken clopidogrel [75 mg/day] for more than 72 hours, no loading dose was needed). Anticoagulation with heparin during the procedure was administered according to the protocol recommendations. Dual antiplatelet therapy with aspirin (100 mg/day) and clopidogrel (75 mg/day) was continued for at least 12 months after the index procedure.
STUDY ENDPOINTS, DEFINITIONS AND FOLLOW-UP
The primary endpoint of the study was in-stent late lumen loss (LLL) at nine months after the index procedure. Secondary procedure-related endpoints were as follows: (1) device, lesion, and clinical success rates (device success was defined as the attainment of <50% residual stenosis of the target lesion using only the assigned device; lesion success was defined as the attainment of <50% residual stenosis, TIMI 3 flow, no residual dissection and thrombosis of the target lesion using any percutaneous method; clinical success was defined as attainment of lesion success of the target lesion and no in-hospital major adverse cardiac event); (2) in-stent and in-segment binary restenosis rates, in-segment LLL, in-stent and in-segment percentage of diameter stenosis; (3) target lesion failure (TLF, device-oriented endpoint), defined as the composite of cardiac death, target vessel myocardial infarction (MI), or ischaemia-driven target lesion revascularisation (iTLR) at one month, six months, 12 months, and annually up to five-year follow-up; (4) the patient-oriented composite of all-cause death, all MI, or any revascularisation at one month, six months, 12 months, and annually up to five-year follow-up; (5) definite and probable ST according to ARC definitions (early, late, and very late)15.
QUANTITATIVE CORONARY ANGIOGRAPHY
Quantitative coronary angiography (QCA) analysis was performed at baseline and nine-month follow-up. All angiograms were evaluated by an independent angiographic core laboratory (China Cardiovascular Research Foundation [CCRF], Beijing, China) using the QAngio XA Version 7.2 Analysis Software (Medis Medical Imaging System Inc., Leiden, The Netherlands). Standard QCA methodology was used including analysis of the stent and the peri-stent segments of 5 mm proximal and distal to the stent edge16. Binary restenosis was defined in every segment (5 mm proximal, 5 mm distal and in-stent) as a >50% diameter stenosis at follow-up. LLL was defined as the difference between the post-procedure and follow-up minimal lumen diameter (MLD).
STATISTICAL METHODS
The sample size estimation was based on a non-inferiority test for the primary endpoint of in-stent LLL at nine-month follow-up after the index procedure. The published angiographic results of the XIENCE V stent in the SPIRIT III trial showed an in-stent LLL of 0.16±0.41 mm at eight months17. Assuming an anticipated in-stent LLL of the FIREHAWK stent of 0.17 mm at nine months and a non-inferiority margin of 0.13 mm, a two-sided alpha of 0.05 and 80% statistical power would require a minimum number of 368 subjects (184 subjects per group). Assuming a loss to angiographic follow-up rate of 20%, a total sample size of 460 enrolled patients was required.
Continuous variables are expressed as mean±standard deviation (SD). Categorical variables are described by counts and percentages. The Student’s t-test and chi-square test/Fisher’s exact test were used to assess the homogeneity of demographic variables and baseline lesion characteristics. The ANCOVA analysis was used as the primary analysis for in-stent LLL. The pre-specified covariates were post-procedure MLD and clinical centre. After eliminating the interaction between treatment group and clinical centre, the difference of the primary endpoint and its 95% confidence interval were estimated by the least squares estimation. All statistical tests were two-sided, and the significant level was 0.05. All analyses were performed with the SAS 9.13 software (SAS Institute Inc., Cary, NC, USA).
Results
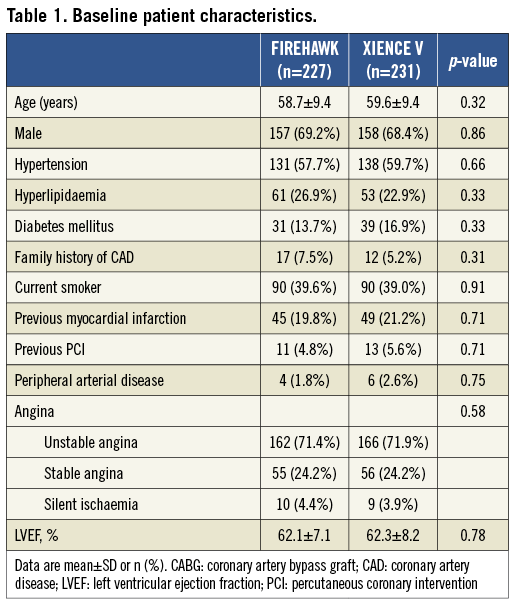
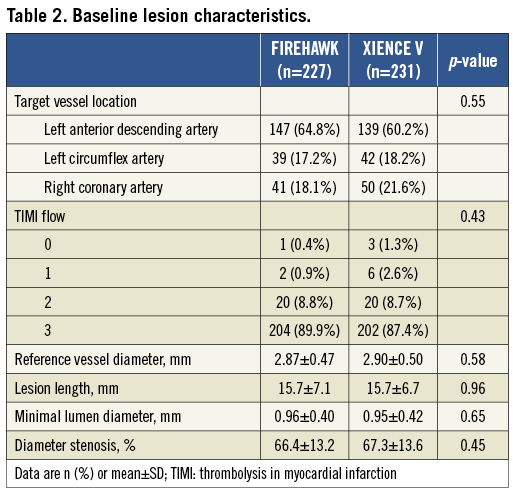
PATIENT, LESION AND PROCEDURAL CHARACTERISTICS
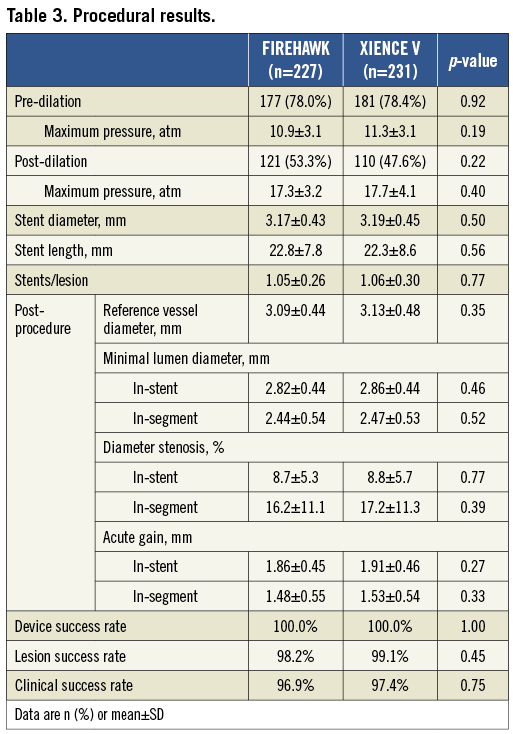
In total, 458 patients were enrolled at 16 Chinese sites and randomised to receive the FIREHAWK (n=227) or the XIENCE V (n=231) stent. Baseline characteristics of the patients and lesions were well matched between the two groups (Figure 2, Table 1 and Table 2). The in-stent % diameter stenosis and in-stent acute gain of the FIREHAWK group after PCI were similar compared to the XIENCE V group (8.7±5.3% vs. 8.8±5.7%, p=0.77; 1.86±0.45 mm vs. 1.91±0.46 mm, p=0.27; respectively). Device success rate was 100.0% in both groups; lesion success rate was 98.2% in the FIREHAWK group and 99.1% in the XIENCE V group, respectively (Table 3).
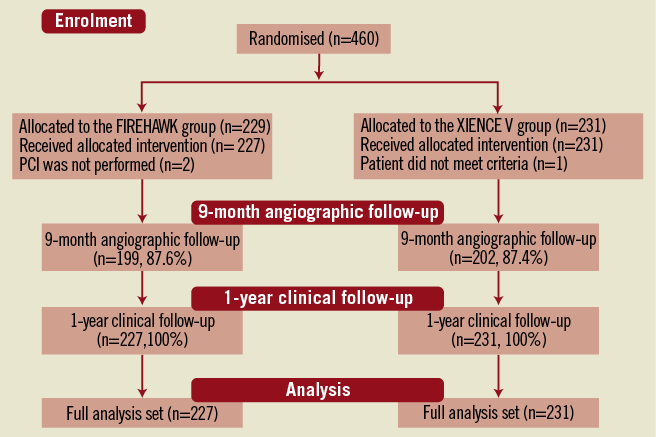
Figure 2. Patient flow.
QUANTITATIVE CORONARY ANGIOGRAPHY ANALYSIS AT NINE-MONTH FOLLOW-UP
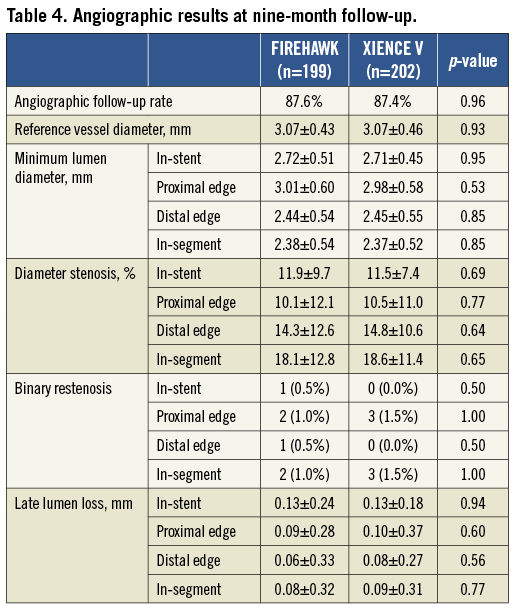
Angiographic follow-up at nine months was completed in 87.6% (199/227) of the FIREHAWK group and 87.4% (202/231) of the XIENCE V group. The primary endpoint of in-stent LLL in the FIREHAWK group was non-inferior to that in the XIENCE V group (0.13±0.24 mm vs. 0.13±0.18 mm, p=0.94; difference and 95% confidence interval 0.00 [–0.04, 0.04] mm; p for non-inferiority <0.0001) (Figure 3, Table 4).
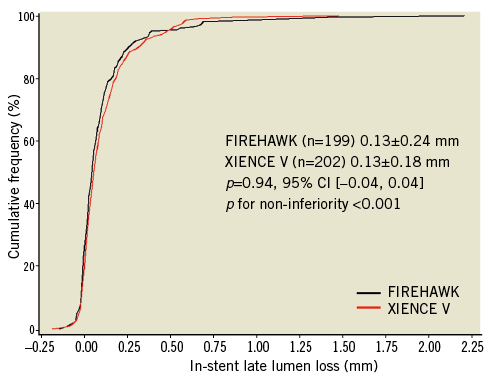
Figure 3. In-stent late lumen loss distribution at nine-month follow-up. CI: confidence interval
CLINICAL OUTCOMES
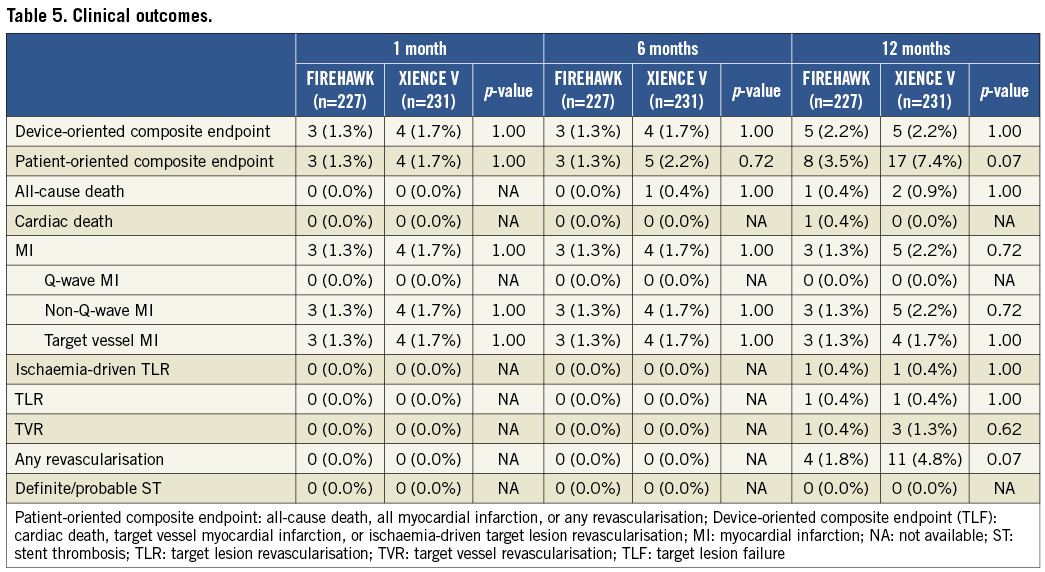
Major clinical endpoint results are summarised in Table 5. The individual and composite of clinical outcome rates were low and comparable in both groups at one, six and 12-month follow-up. However, the FIREHAWK-treated group showed a non-significant trend towards a low rate of patient-oriented clinical outcomes compared with the XIENCE V group (3.5% vs. 7.4%, p=0.07, Table 5). During the 12-month follow-up, no definite/probable ST was observed.
Discussion
This prospective, multicentre, randomised trial has demonstrated for the first time that: 1) the novel biodegradable polymer SES (FIREHAWK) has similar safety and efficacy for the treatment of patients with de novo lesions compared to the EES (XIENCE V); 2) in comparison with the durable polymer EES, biodegradable polymer SES shows non-inferiority of antirestenotic efficacy with respect to the primary endpoint of in-stent LLL; 3) the clinical outcomes were low in both the biodegradable polymer SES and durable polymer EES groups, and no ST was observed at one-year follow-up; 4) the concept of sirolimus target release using a novel abluminal groove-filled biodegradable PLA polymer is feasible and effective compared to the contemporary proven EES.
BIODEGRADABLE POLYMER SES WITH UNIQUE DESIGN
The novel biodegradable polymer SES investigated in the present study was described previously13, consisting of three components: a cobalt-chromium L605 platform, biodegradable PLA polymer and the antiproliferative drug sirolimus. In particular, biodegradable polymer localised in the abluminal grooves of the struts is fully metabolised to water and carbon dioxide within six to nine months. This unique design with abluminal groove-filled biodegradable polymer is completely different from other biodegradable polymer DESs with a laser-cut reservoir design (such as CoStar [Conor MedSvastems, Menlo Park, CA, USA], NEVO [Cordis Corporation, Bridgewater, NJ, USA])18-20. Meanwhile, the unique groove-filled biodegradable polymer design with minimally contacting intima potentially might be associated with a decrease of polymer-related inflammation. In addition, a finite element analysis showed that the stresses and deformations of the device during expansion are only focused on the hinge points of the stent13. Therefore, the impact on the integrity of biodegradable polymer poured into the abluminal grooves was reduced in patients undergoing PCI.
The clinical efficacy of DES mainly relates to stent design, active drug and the presence and type of polymer. Though sirolimus and everolimus were used in the stents in this current study, Steigerwald and colleagues proved that these mTOR inhibitors have similar effects on endothelial regrowth and neointimal thickening21. In addition, it should be noted that the average sirolimus dosage of the FIREHAWK is only 3 µg/mm stent length compared to 100 µg/mm2 of stent surface area in the XIENCE V stent13. Given the similar and low in-stent LLL, sufficient inhibition of neointimal proliferation may have been achieved in the FIREHAWK stent using the groove-filled biodegradable polymer design compared with the XIENCE V. Based on this unique technique, the targeted drug release may potentially result in comparable drug release kinetics and uptake of the drug into the arterial wall. Therefore, it appears clear that both stent systems in this study contributed to the comparable efficacy in neointimal suppression.
EFFICACY OF THE NOVEL BIODEGRADABLE POLYMER SES
Numerous studies have shown that in-stent LLL is a predictor of future cardiovascular events. It has been used as a surrogate endpoint to compare different devices, though the causes of restenosis after revascularisation were multifactorial22-24. In permanent metallic stent platforms, the LLL is solely due to neointimal proliferation, and thus the LLL provides an indirect angiographic evaluation of the vessel wall response to the endovascular device25. In a small randomised study (SPIRIT II), the in-stent LLL was 0.17±0.32 mm at six months, and delayed neointimal hyperplasia was observed with an in-stent LLL of 0.33±0.37 mm at two years26. However, the biodegradable polymer SES with only a metallic stent left after six to nine months post-procedure was potentially associated with a minimal inflammation and hypersensitivity response compared with durable polymer DES after polymer biodegraded in long-term follow-up27,28. In addition, though a fully bioresorbable scaffold (BRS) may be the ideal device for treatment with PCI, there are no data currently available for direct comparison of the efficacy of the devices between BRS and DES29.
Previous studies showed that employing a biodegradable coating on a stent platform led to superior clinical and angiographic outcomes7,30-32. The adverse clinical events of this study were markedly low in both stent groups of patients with de novo lesions compared to other studies. It may be partly due to the highly effective biodegradable polymer SES and EES used in the present study. Alternatively, the strictly selected population of this study may have contributed to the low incidences of adverse clinical events. These observations at least indicated a greater degree of neointimal hyperplasia suppression in both devices. However, it is hard to reach a statistical conclusion regarding the clinical outcomes between these two devices due to the low incidence of events. Recently, the COMPARE II randomised controlled trial showed that a biodegradable polymer biolimus-eluting stent is as safe and efficacious as the EES33. However, the present study confirmed for the first time the non-inferiority of the novel biodegradable polymer SES compared to EES with respect to nine-month in-stent LLL. The underlying causes of non-inferiority are potentially attributed to the unique design and biodegradable polymer.
DEVICE-RELATED STENT THROMBOSIS
Stent thrombosis is one of the most prominent concerns with the widespread use of DES in daily clinical practice34,35. The newer-generation DES, including EES, have been developed with the intention of improving the overall safety of earlier DES while maintaining antirestenotic efficacy. A meta-analysis from 13 randomised trials suggests a benefit of EES in reducing ST36. Though the cause of this detrimental event is more likely to be multifactorial, delayed re-endothelialisation and hypersensitivity induced by durable polymer play an important role.
In the present study, ST at one-year follow-up was not observed in either group. Therefore, it is difficult to provide a powerful comparison with respect to this problem. Long-term follow-up of the TARGET I trial and the real-world TARGET II study evaluating the FIREHAWK stent in a broad patient population is necessary before definite conclusions can be drawn concerning this important clinical outcome.
STUDY LIMITATIONS
This study has several limitations. First, the sample size was calculated to assess a difference in LLL, which is a surrogate endpoint for clinical restenosis. Secondly, patients included in the study were selected strictly by angiographic and clinical characteristics rather than as in an all-comers trial; thus, the results may not be regarded as being applicable to routine clinical practice. Thirdly, the present study did not include additional intracoronary imaging guidance and examinations (such as intravascular ultrasound, optical coherence tomography). Finally, in the early stages the trial comparing DES could not provide a direct assessment of the effect of different polymers and drug used, which could have modulated the final performance of the stents9,37,38. Given the aforementioned limitations, a large-scale randomised trial conducted on less stringently selected and higher risk patients is warranted to determine the long-term efficacy and safety of this novel target release sirolimus-eluting coronary stent with biodegradable polymer in the abluminal grooves.
Conclusions
The TARGET I trial demonstrates that the novel abluminal groove-filled biodegradable polymer sirolimus-eluting stent has similar efficacy and safety compared to the contemporary, widely used durable polymer everolimus-eluting stent for patients with single de novo coronary lesions.
Acknowledgements
We appreciate the great effort of the clinical research coordinators in the TARGET I study organisation and participating centres listed in the Appendix.
Funding
This study was sponsored by MicroPort Scientific Corporation, Shanghai, China.
Conflict of interest statement
R-L. Gao has received research grants from MicroPort, Abbott Vascular, B. Braun and Boston Scientific. B. Xu is a consultant for MicroPort. The other authors have no conflicts of interest to declare.
Online appendix
TARGET I STUDY ORGANISATION
Principal Investigators: Run-Lin Gao, Martin B. Leon
Executive Committee: Run-Lin Gao, Martin B. Leon, Bo Xu
Angiographic Core Lab: CCRF, Beijing, China (Consultant: Alexandra J. Lansky)
Clinical Events Committee: Yong Huo (Chair), Weimin Wang, Wei Gao, Lefeng Wang
Data Management and Statistics: Wei Li, Yang Wang, Xuan Jia, Division of Biometrics, National Center for Cardiovascular Diseases of China
Data Monitoring and Co-ordination: CCRF, Beijing, China (Qi-Chao Zhai, Ye-Lin Zhao)
COLLABORATORS
Fu Wai Hospital, National Center for Cardiovascular Diseases of China, Beijing (Jue Chen, Ke-Fei Dou, Li-Jian Gao, Chong-Jian Li, Hai-Bo Liu, Chao-Wei Mu, Jie Qian, Shu-Bin Qiao, Xue-Wen Qin, Hong Qiu, Yong-Jian Wu, Liang Xu, Hong-Bing Yan, Yue-Jin Yang, Shi-Jie You).
Affiliated Anzhen Hospital of Capital Medical University, Beijing (Li-Ying Chen, Yu-Tong Cheng, De’an Jia, Zhi-Zhong Li, Wen-Xian Liu, Xin-Min Liu, Yu-Yang Liu, Shu-Zheng Lu, Chang-Sheng Ma, Bin Nie, Shang-Qiu Ning, Dong-Mei Shi, Kai Tan, Feng Xu, Dong-Hua Zhang, Yu-Jie Zhou).
Shenyang Northern Hospital, Shenyang (Ya-Ling Han, Quan-Min Jing, Hai-Wei Liu, Ying-Yan Ma, Bin Wang, Geng Wang, Xiao-Zeng Wang, Kai Xu).
Affiliated Nanjing First Hospital of Nanjing Medical University, Nanjing (Shao-Liang Chen, Zhi-Zhong Liu, Ling Lin, Song Lin, Shou-Jie Shan, Hai-Mei Xu, Fei Ye, Wei You, Jun-Jie Zhang, Zhong-Sheng Zhu).
Daqing Oil Field General Hospital, Daqing (Hui Li, Sheng-Quan Liu, Zhi-Qi Sun, Bai-Ying Wang, Shang-Yu Wen).
Affiliated Ruijin Hospital of Shanghai Jiaotong University School of Medicine, Shanghai (Qin Chen, Feng-Hua Ding, Run Du, Jian Hu, Wei-Feng Shen, Zhen-Kun Yang, Jian-Sheng Zhang, Qi Zhang, Rui-Yan Zhang).
Affiliated SRRS Hospital of Zhejiang University School of Medicine, Hangzhou (Guo-Sheng Fu, He Huang, Zhan-Lu Li, Shao-Xiang Weng).
First Affiliated Hospital of Xi’an Jiaotong University School of Medicine, Xi’an (Xiao-Jun Bai, Ning Guo, Hong-Bing Li, Xin-Jun Lei, Jian-Jun Mou, Dong-Qi Wang, Zu-Yi Yuan).
Wuhan University People’s Hospital, Wuhan (Jing Bai, Xiao-Rong Hu, Hong Jiang, Xue-Jun Jiang, Bo Yang).
Liaoning Provincial People’s Hospital, Shenyang (Cheng-Yang Li, Zhan-Quan Li, Li Liu, Yun-Qi Shi, Yong-Xin Wang, Long Yuan).
First Affiliated Hospital of Harbin Medical University, Harbin (Guo Dong, Wei-Min Li, Yue Li, Dang-Hui Sun, Jian-Tao Sun).
First Central Hospital of Tianjin, Tianjin (Jing-Yi Liang, Ying Liu, Cheng-Zhi Lu, Da-Sheng Xia, Jian-Qiang Xu, Xiang-Dong Zhao).
Guangdong Provincial People’s Hospital, Guangzhou (Ji-Yan Chen, Zhu-Jun Chen, Peng-Cheng He, Wen-Hui Huang, Yuan Liu, Ning Tan, Nian-Jin Xie, Dan-Qing Yu).
Affiliated Zhongshan Hospital of Fudan University, Shanghai (Jun-Bo Ge, Lei Ge, Dong Huang, Xue-Bo Liu, Ju-Ying Qian, Kang Yao, Feng Zhang, Xin Zhong).
Chinese PLA General Hospital, Beijing (Jun Chen, Yun-Dai Chen, Kai Guo, Chang-Fu Liu, Hong-Bin Liu, Zhi-Jun Sun).
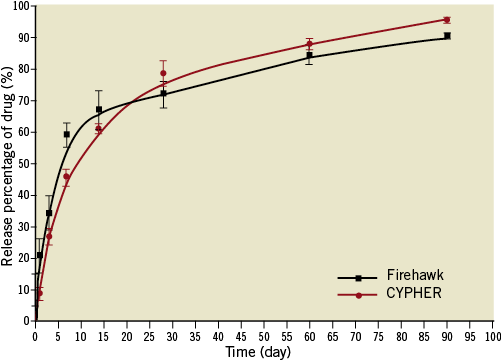
Online Figure 1. The drug release kinetics. Preclinical study showed that the drug release kinetics curves were similar between the Firehawk and CYPHER stents.
