Abstract
Aims: To assess the two-year clinical follow-up of the NEVO RES-1 study, a randomised comparison between the NEVO™ sirolimus-eluting coronary stent system (NEVO SES) and the TAXUS Liberté™ paclitaxel-eluting stent (TAXUS PES).
Methods and results: NEVO RES-I randomised 394 patients with single de novo lesions with a maximum length of 28 mm and diameter of 2.5-3.5 mm to NEVO SES (n=202) versus TAXUS PES (n=192). Six-month angiographic results demonstrated the superiority of the NEVO SES over the TAXUS PES for the primary endpoint, in-stent late loss. At one year, MACE (death, emergent CABG, TLR, and MI) in the NEVO SES group was 6.1% versus 10.6% in the TAXUS PES group (p=0.139). After two years, MACE was 7.2% in the NEVO SES group versus 13.0% in TAXUS PES group (p=0.086). Corresponding rates of TLR were 3.6% versus 7.6% (p=0.116). No ARC-defined definite or probable stent thromboses (ST) were reported with NEVO SES while two occurred with TAXUS PES.
Conclusions: While not designed or powered for clinical endpoints, individual and composite clinical endpoints numerically favoured the NEVO SES over the TAXUS PES, with continued separation over time up to two years. No ARC-defined definite or probable ST was reported in the NEVO SES group at two years. Clinical trial identifier: NCT00606333 http://www.clinicaltrials.gov
Introduction
The NEVO™ sirolimus-eluting coronary stent (NEVO SES) (Cordis Corporation, Bridgewater, NJ, USA) is a cobalt-chromium alloy stent with a platform that incorporates reservoir technology and a bioabsorbable polymer from which sirolimus is released. Multiple laser-cut reservoirs are filled with a blend of sirolimus and the bioabsorbable polymer, substantially reducing the amount of tissue exposed to the polymer. Moreover, absorption of the polymer occurs within approximately three months, further limiting the duration of vessel wall exposure to the polymer. Thereafter, only a biologically inert bare metal platform remains.
The NEVO RES-I study is a prospective multicentre, randomised, non-inferiority comparison with the TAXUS Liberté™ paclitaxel-eluting coronary stent (TAXUS PES) (Boston Scientific Corp., Natick, MA, USA) combining an angiographic primary endpoint with collation of clinical data. The six-month quantitative coronary angiographic (QCA) results demonstrated an in-stent late loss of 0.13±0.31 mm in the NEVO SES group versus 0.36±0.48 mm in the TAXUS PES group, a difference that met predefined criteria for both non-inferiority and superiority (p<0.001)1. A 100-patient intravascular ultrasound (IVUS) pre-specified substudy showed more uniform and significantly greater suppression of neointimal hyperplasia with less positive remodelling in the NEVO SES group versus the TAXUS PES group2,3.
Whilst the NEVO RES-I trial was not designed or powered to show statistically significant differences in individual or composite clinical outcomes, all clinical endpoints showed differences at six months which numerically favoured the NEVO SES1. Clinical follow-up is planned to continue up to five years. We report the clinical outcomes to two years following the index procedure.
Methods
Full details of the NEVO RES-I trial design have been published previously1. Briefly, patients ≥18 years of age at 40 medical centres in nine countries were randomly assigned in a single-blind 1:1 design to undergo implantation of NEVO SES versus TAXUS PES. Patients were eligible if they presented with a left ventricular ejection fraction >30%, stable or unstable angina or silent ischaemia, and a single >50% and <100% de novo stenosis up to 28 mm in length in a native coronary artery 2.5-3.5 mm in diameter. Angiographic exclusion criteria included left main coronary artery disease, bifurcation lesions, ostial lesions, chronic total occlusions, and lesions with severe calcification or excessive tortuosity. Major clinical exclusion criteria included a PCI within the preceding 30 days, acute myocardial infarction (MI) less than72 hours, creatinine >2.0 mg/dl, left ventricular ejection fraction ≤30% or other significant comorbidities. Randomisation was performed during the PCI procedure, after the guidewire was passed.
Patients were randomly assigned to study stents on a 1:1 basis within each enrolling medical centre and further stratified by diabetic versus non-diabetic status to prevent imbalance between the two groups. Periprocedural anticoagulation with heparin or bivalirudin and glycoprotein IIb/IIIa inhibitors was administered according to local practices. All patients were treated with aspirin or clopidogrel before undergoing the index PCI; dual antiplatelet therapy with 75 mg of clopidogrel and aspirin ≥75 mg daily was mandated for ≥6 months after the procedure, with aspirin to be continued indefinitely.
Endpoints of the trial
The primary endpoint of the NEVO RES-I trial was in-stent late luminal loss (LLL) at six months, ascertained by core lab QCA. Prespecified, secondary clinical endpoints of the trial included: 1) target lesion (TLR) and target vessel (TVR) revascularisation; 2) target lesion failure (TLF), a composite of a) cardiac death that could not be attributed to a non-cardiac event or to a vessel other than the target vessel, b) target vessel-related MI, and c) clinically-driven TLR; 3) target vessel failure (TVF), which included any TVR, MI or cardiac death that could not be attributed to a non-target vessel; 4) major adverse cardiac events (MACE), a composite of a) cardiac or non-cardiac death, b) emergent CABG surgery, c) Q- or non-Q-wave myocardial infarction (MI), defined by the World Health Organisation on the basis of creatine kinase and creatine kinase-MB enzyme rise4, and d) target lesion revascularisation (TLR); 5) stent thrombosis (ST), applying the definitions of the Academic Research Consortium5 at hospital discharge, 30 days, six months and then annually up to five years post index procedure.
Prior to the scheduled or unscheduled coronary angiographies, the protocol mandated the investigators to document ischaemia. Revascularisation of the target vessel or lesion was defined as clinically driven in case of a positive functional ischaemia study or ischaemic symptoms and an angiographic minimal lumen diameter stenosis ≥50% by quantitative coronary angiography (QCA), or if revascularisation was performed on a target vessel with a diameter stenosis ≥70%.
Patient follow-up and data collection
All patients underwent angiographic follow-up at six months and ambulatory follow-up visits at 30 days, six months, one year and two years. Patient data were collected via electronic case report forms (CRF) (KIKA Medical, Paris, France) and automatically transferred to an independent clinical research organisation (CRO) (Harvard Clinical Research Institute, Boston, MA, USA) for data management and analysis. Study monitors performed 100% source data verification on-site. An independent and blinded clinical events committee (CEC) adjudicated all deaths, MI, ST, revascularisation procedures and cerebrovascular events.
Statistical analysis
A sample size of 388 patients was estimated to test the non-inferiority or superiority of the NEVO SES compared with the TAXUS PES in the analysis of the primary endpoint, as described previously1. This study was not powered to test the effects of stent allocation on clinical endpoints such as death, TLR, MI and ST. Subgroup analyses were pre-specified for diabetic versus non-diabetic status, lesions ≤ versus >20 mm in length, and for single vs. overlapping stents. The data were analysed according to the stent to which the patient was randomised (i.e., on an intention-to-treat basis). Continuous variables are reported as mean ± standard deviation, and discrete variables as counts and percentages. Continuous variables were analysed using Student’s t-tests and discrete variables were analysed using Fisher’s exact test. A p-value of <0.05 was considered significant. The statistical analyses were performed using the SAS® statistical system for Windows (SAS Institute Inc., Cary, NC, USA) version 9.1 or higher. While pre-specified, individual and composite endpoints compared after six months are for descriptive purposes only; no corrections have been made for multiple comparisons. Revascularisation of the target lesion and MACE during follow-up were analysed by the Kaplan-Meier method. The differences between the event-free survivals were compared by Wilkinson and log-rank tests.
Results
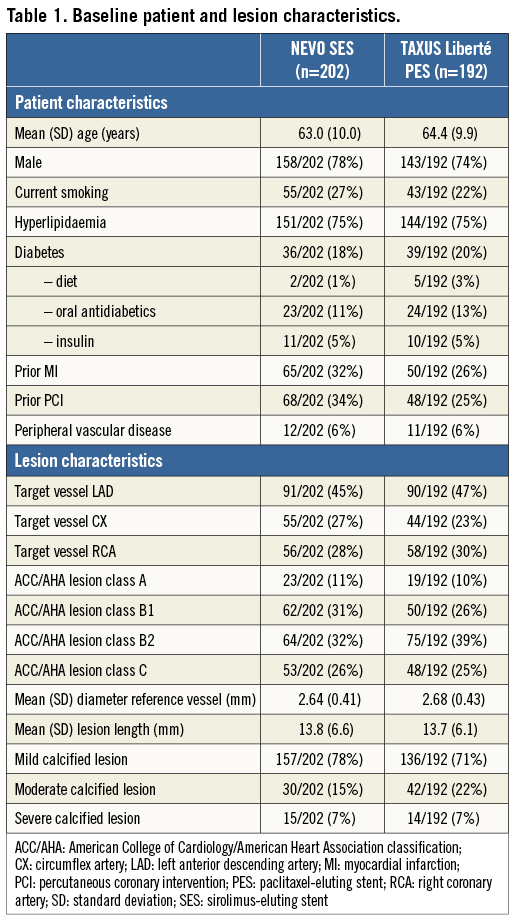
Between March and October 2008, 394 patients were enrolled in the NEVO RES-I trial, of whom 202 were randomly assigned to NEVO SES and 192 to TAXUS PES implantations. The baseline patient and lesion characteristics are shown in Table 1. The primary endpoint of in-stent late luminal loss at six months after the index procedure demonstrated superiority of the NEVO SES over the TAXUS PES (0.13±0.31 mm vs. 0.36±0.48 mm, p<0.001 for both non-inferiority and superiority). Device success (defined as obtaining a <50% residual stenosis with the assigned device) was 99.5% in both groups1. Dislodgement of a NEVO stent outside of the coronary arteries occurred in one patient when the operator tried to retrieve the device after a failure to cross. This patient was then successfully treated with two NEVO stents.
Clinical outcomes accumulated at two years
At two years, clinical data compliance was 96.0% and 96.4% for the NEVO SES and TAXUS PES arms, respectively (Figure 1). Clinical outcomes are shown in Table 2. Event rates remained consistently lower for NEVO SES than for TAXUS PES at two years of follow-up. While the rates of in-hospital events were nearly identical in both groups, differences in out-of-hospital events (reflecting differences in outcomes more likely due to the stent rather than the procedure) increased during the two-year follow-up. The differences in out-of-hospital MACE rate increased and became significant at two years. The individual components of MACE each numerically favoured NEVO SES over TAXUS PES although the differences were not statistically significant.
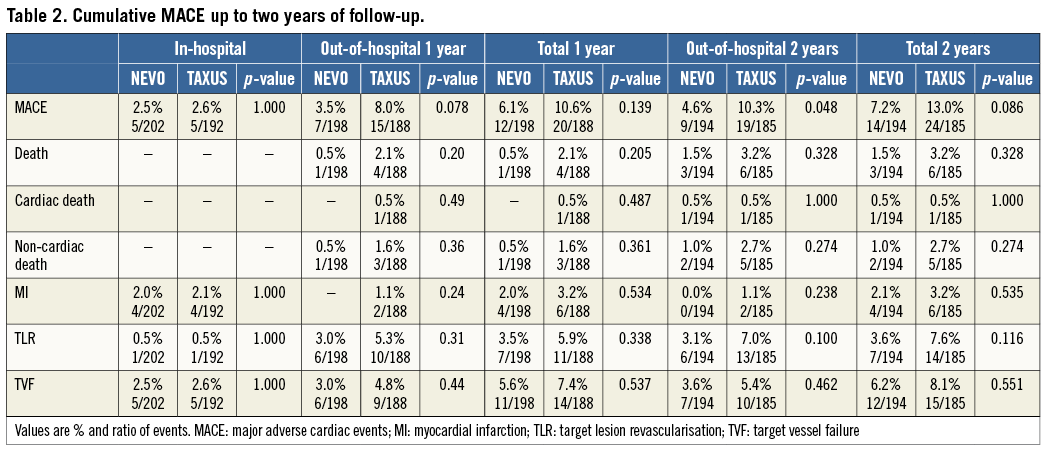
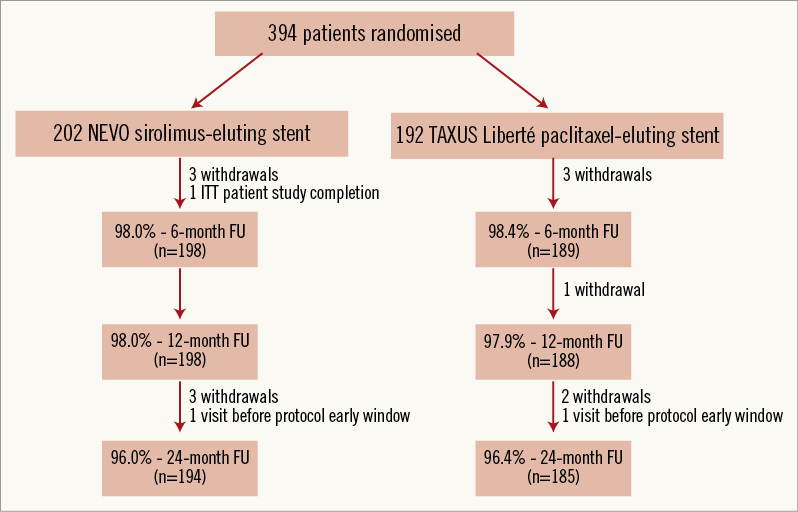
Figure 1. Patient flow chart with follow-up compliance. ITT: intention to treat; FU: follow-up
Kaplan-Meier curves of survival free from MACE and from TLR are shown in Figure 2. Differences in survival free from MACE for NEVO SES versus TAXUS PES increased from 96.0 vs. 92.1% (p=0.108) at six months, to 94.5% vs. 90.0% (p=0.106) at one year and to 93.0% vs. 87.3% (p=0.062) at two years. Differences in survival free from TLR increased from 98.5% vs. 96.3% (p=0.170) at six months, to 96.5% vs. 94.1% (p=0.268) at one year, to 96.5% vs. 92.4% (p=0.087) at two years.
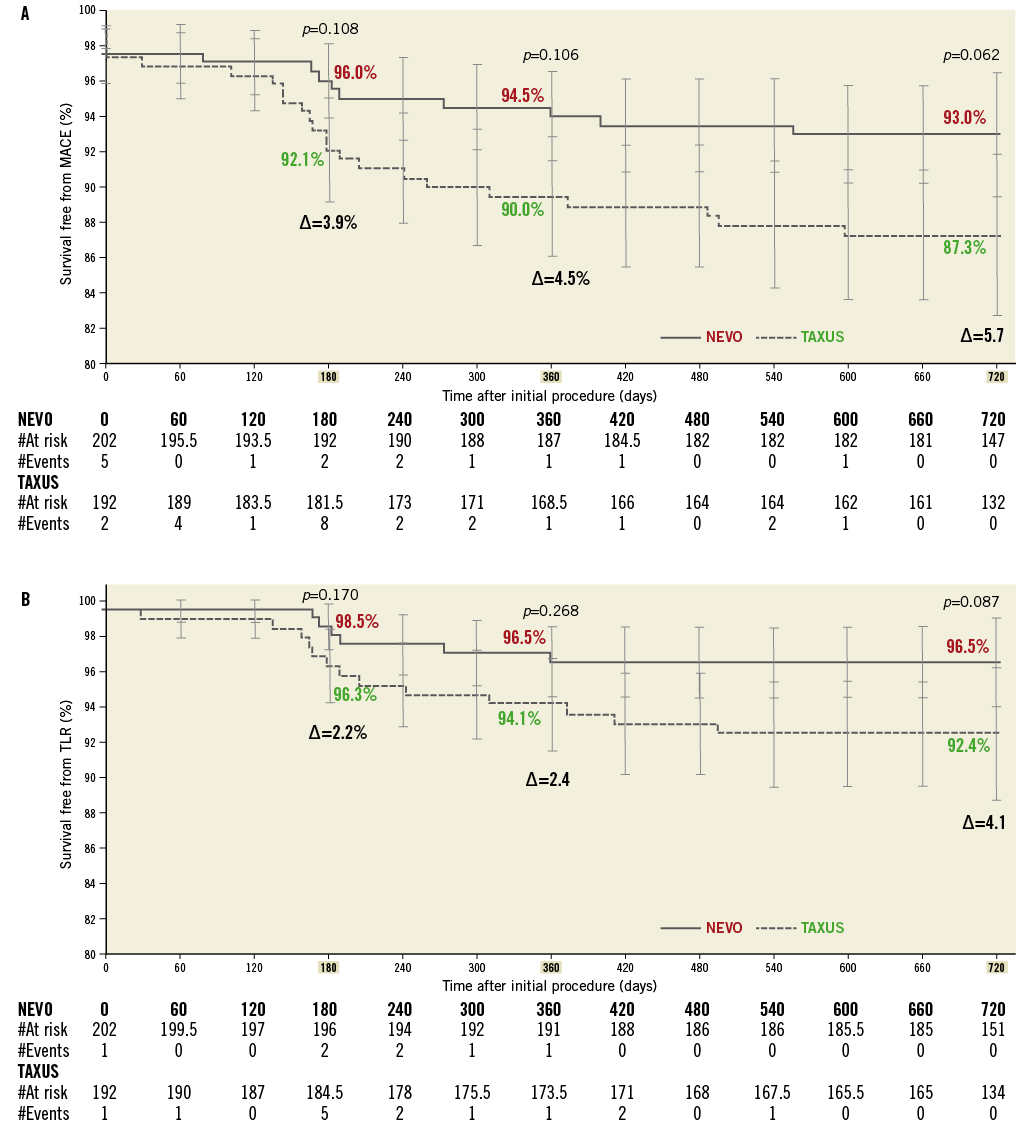
Figure 2. A) Kaplan-Meier* curves of freedom from MACE to 24 months. B) Kaplan-Meier* curves of freedom from TLR to 24 months. *Kaplan-Meier estimates are based on ITT patients, where patients not experiencing the event are censored at 720-day or last known follow-up, whichever is earlier. MACE: major adverse cardiac events; TLR: target lesion revascularisation
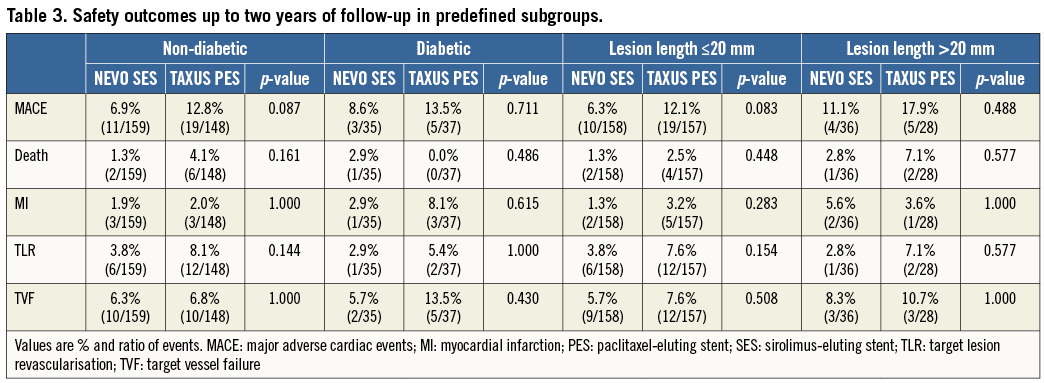
In the prespecified subgroups, the same differences in favour of NEVO SES were noted (Table 3).
Clinical outcomes between six months and two years
Between six months and two years after the index procedure, two additional deaths occurred in the NEVO SES group: one cardiac death at day 555 (unknown death which was adjudicated as a cardiac death) and one non-cardiac death (cancer). Three non-cardiac deaths were recorded in the TAXUS PES group (cancer, Parkinson’s disease, non-haemorrhagic stroke). In the NEVO SES group no MI occurred, versus one MI (non-Q-wave, target vessel-related) in the TAXUS PES group at day 190.
No ARC-defined probable or definite stent thromboses were observed during the two-year follow-up in the NEVO SES group. In the TAXUS PES group, one definite stent thrombosis occurred at day 410 in the TAXUS PES group in a patient who had previously presented with a probable stent thrombosis at day 180. In both cases, the patient was on aspirin and clopidogrel at the time of the stent thrombosis event.
Four TLRs occurred in the NEVO SES group and seven in the TAXUS PES group after six months. Respective days after index and restenosis pattern according to the Mehran classification6 are: day 189/ISR 2, day 182/ISR 1B, day 273/ISR 3, day 359/ISR 1C for NEVO SES and day 241/ISR 1C, day 189/ISR 1C, day 204/ISR 1C, day 495/ISR 1B, day 372/ISR 0, day 309/ISR 1B and day 410/ISR 4 for TAXUS PES TLRs. All TLRs and TVRs were clinically driven, according to the protocol definition.
Medication - antiplatelet use
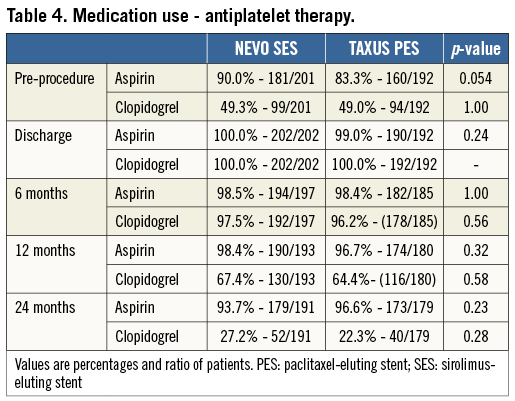
Overall, rates of aspirin and clopidogrel use were similar during the two years of follow-up (Table 4). At discharge, 100% of NEVO SES and 99% of TAXUS PES patients were on aspirin and clopidogrel. Aspirin usage remained high (>90%) up to two years in all patients. Clopidogrel use remained high up to six months of follow-up (>98%) as recommended in the study protocol but then decreased to <70% at one year and further decreased to <30% at two years.
Discussion
The NEVO RES-I study assessed the NEVO SES that releases sirolimus from a local drug-delivery platform concentrating a bioabsorbable polymer-drug blend in dedicated reservoirs instead of using a surface coating. While the NEVO RES-I trial was not powered to demonstrate significant differences in clinical outcomes, the two-year follow-up showed numerically fewer adverse events in the NEVO SES compared to the TAXUS PES group, which increased over time. No definite or probable stent thrombosis was noted in the NEVO SES group at two years. Interestingly, numerical differences favouring the NEVO SES that were observed at six months continued to increase over the 24 months of follow-up. Fewer events occurred in the NEVO SES arm for both individual as well as the composite clinical endpoints that included death, MI, and TLR. No differences were observed in event rates from index procedure to hospital discharge, reflecting similar safety outcomes in the periprocedural period. The observed differences were most notable in the clinical events that occurred following hospital discharge.
The net clinical value of DES, compared with bare metal stents, lies in the degree to which they reduce the clinical burden of restenosis, offset by a possible elevation in the risk of ST. Efforts to design stents that will optimise this net clinical value are warranted. NEVO ResElution-I was the first trial to assess a new local drug-delivery platform concentrating a bioabsorbable polymer-drug blend in dedicated reservoirs instead of using a surface coating. This design may remove a major possible mechanism of DES ST, namely the exposure of the coronary tissue to potentially proinflammatory polymer material.
Several trials have assessed stent designs incorporating biodegradable polymers. The long-term results are conflicting. The LEADERS trial randomly compared in 1,707 patients a stainless steel stent eluting Biolimus-A9 (BES) from an abluminal biodegradable polylactic acid polymer (BioMatrix Flex™; Biosensors Inc., Newport Beach, CA, USA) to the Cypher Select™ SES (Cordis, Miami Lakes, FL, USA). The biodegradable polymer metabolises in water and carbon dioxide in six to nine months. At one and four years, BES was non-inferior to Cypher Select SES for the primary endpoint, a composite of cardiac death, MI and clinically indicated TLR. At four years, the risk of cardiac events associated with very late ST was reduced7,8. The ISAR-TEST-3 study compared biodegradable polymer (BP) and polymer-free (PF) SES with the Cypher permanent polymer SES. A total of 605 patients were enrolled. The primary endpoint, LLL at six-month to eight-month angiographic follow-up was 0.17±0.45 mm in the BP stent group, 0.23±0.46 mm in the PF cohort (p<0.001 for non-inferiority)9. At two years, there was no significant difference in TLR, death/MI or definite/probable ST10. Delayed LLL was significantly higher in the PF group. The ISAR-TEST-4 compared biodegradable polymer SES with durable polymer SES and durable polymer everolimus-eluting stents in 2,603 patients11. At three years, there was no significant difference between biodegradable polymer and permanent polymer DES with regard to the primary endpoint, a composite of composite of cardiac death, target vessel-related MI, and TLR. Rates of definite/probable stent thrombosis were also similar in both groups12.
More recently, a stent with abluminal reservoirs has been tested in a randomised trial. The Cre8™ stent (CID SpA, Saluggia, Italy) is characterised by a permanent biocompatible i-Carbofilm™ strut coating and abluminal reservoirs loaded with a polymer-free sirolimus formulation (Amphilimus™). A randomised trial compared the Cre8 stent to TAXUS PES in a total of 323 patients. In-stent late loss was significantly lower with Amphilimus-eluting stents (0.14±0.36 mm versus 0.34±0.40 mm, p<0.0001 for non-inferiority, p<0.0001 for superiority). Definite/probable ST occurred in 0.6% of patients in each group13. Long-term safety and efficacy data are lacking since currently available clinical follow-up is limited to one year. It therefore remains to be determined if delayed LLL and TLR will occur as previously noted with other polymer-free stents10.
Assessment of ST rates is difficult and requires long-term follow-up on large cohorts of patients14,15. However, the absence of definite or probable stent thrombosis in the NEVO SES arm at two years suggests a good mid-term and long-term safety profile of the stent in which the bioabsorbable polymer is restricted to the interior of the reservoirs, thereby eliminating contact with the arterial vessel wall. Preclinical studies have shown that the polymer is resorbed in approximately three months with complete tissue ingrowth into the reservoirs1. These distinctive design features of the NEVO SES may limit temporal and spatial polymer-induced arterial wall inflammation, reducing the risk of ST and the requirement for prolonged dual antiplatelet therapy.
In our follow-up, the superior clinical benefit of NEVO SES was seen in the pre-specified group of diabetic patients. The relative efficacy of different DES in diabetic patients remains controversial. A heterogeneity analysis of the diabetic subgroup of four randomised trials comparing the CYPHER SES with the Bx VELOCITY™ (Cordis) stent suggested a higher rate of adverse events in patients receiving the CYPHER SES, although this has not been confirmed in a large meta-analysis15-17. In trials comparing the XIENCE V everolimus-eluting coronary stent (XIENCE V EES; Abbott Vascular, Redwood City, CA, USA) to the TAXUS SES, outcomes were similar in the diabetic subgroups in contrast to superior outcomes for XIENCE V EES overall and in the non-diabetic population18-20. Paclitaxel has been postulated to be an effective drug in diabetic patients21. For the diabetic subgroup in the present study, in-stent LLL at six months was significantly less for NEVO SES (0.17±0.42 mm) versus TAXUS Liberté PES (0.44±0.55 mm) (p=0.032) and the clinical benefit was maintained at two years, suggesting superior efficacy of NEVO SES in this subgroup.
Limitations
The NEVO ResElution-I trial was designed to assess angiographic differences at six months between groups. Clinical data should therefore be interpreted with caution and is hypothesis-generating. In particular, an increase in TVR and TLR in studies with mandatory angiographic control has been suggested. However, the investigators were required per protocol to document ischaemia before the angiographic procedures. Repeat revascularisations were assessed by an independent CEC and were ischaemia-driven in both groups. Furthermore, differences in TLR continue to increase after the mandated angiographic control at nine months, which clearly supports a more potent inhibition of neointimal proliferation by the NEVO SES.
The TAXUS Liberté PES was chosen as a comparator since it was the most widely used DES when the study was designed. No conclusion on the efficacy of the NEVO SES compared to other DES can be drawn.
NEVO SES was further assessed in the NEVO II study, a randomised comparison with the XIENCE V everolimus-eluting stent. This trial was stopped in October 2010 after enrolment of 156 patients due to stent dislodgement in the NEVO arm. Stent retention was improved and successfully tested in vitro and in animal studies. However, the NEVO programme was discontinued in June 2011. This decision was based on changing business dynamics in the drug-eluting stent market22.
Conclusions
The NEVO SES demonstrated superiority for the primary endpoint of six-month in-stent late loss compared to the TAXUS PES with both lower absolute late loss measurements and more uniform suppression of neointimal hyperplasia. For all measured clinical indices at six months, one year and two years post procedure, event rates were numerically fewer for NEVO SES than for TAXUS PES and the numerical difference observed increased over time. For the composite endpoint, MACE, there was a strong trend for improved outcomes with NEVO for the entire cohort (p=0.086), predominantly driven by lower rates of TLR for NEVO SES. These differences are most notable when clinical events from hospital discharge to longest follow-up are evaluated. No ARC definite or probable stent thromboses were observed in the NEVO SES arm, while two were reported in the TAXUS PES arm. The association of reservoir technology and a bioabsorbable polymer may mitigate thrombosis risk without compromising antirestenotic efficacy.
Acknowledgements
We thank Liesbeth Vanderlinden for her contribution to this manuscript.
Appendix
The following investigators participated in the NEVO RES-I Study:
Steering Committee: Alexandre Abizaid; John A. Ormiston; Jean Fajadet; Laura Mauri; Joachim Schofer; Stefan Verheye; Christian Spaulding
Data and Safety Monitoring Board (HCRI): MichaelFarkouh; David Faxon; Bernard Gersh; Christian Hamm; Gary Mintz; E. John Orav
Clinical Events Committee (HCRI): Cliff Berger; Scott Bortman; Frank Bowen; John Dashe; Laurence Epstein; Eli Gelfand; Satyendra Giri; David Gossman; Allen Hamdan; Thomas Hauser; Joseph Kannam; Kamal Khabbaz; Megan Leary; David Litvak; Warren Manning; Peter Oettgen; Duane Pinto; David Thaler; Sergio Waxman
Clinical Sites: Monash Medical Center, Clayton, Victoria, Australia - Prof. Meredith; St. Vincents Hospital, Melbourne, Australia - Dr. Whitbourn; Royal Adelaide Hospital, Adelaide, Australia - Dr. Worthley; AZ Middelheim, Antwerp, Belgium - Dr. Verheye; Ziekenhuis Oost-Limburg campus Sint-Jan, Genk, Belgium - Prof. Dens; Universitaire Ziekenhuizen Leuven, Leuven, Belgium - Prof. Dubois; CHU Sart Tilman, Liège, Belgium - Prof. Legrand; Onze Lieve Vrouwziekenhuis, Aalst, Belgium - Dr. Wijns; Imelda Ziekenhuis, Bonheiden, Belgium - Dr. Debruyne; Instituto Dante Pazzanese de Cardiologia, Sao Paulo, Brazil - Dr. Abizaid; Incor-Instituto do Coraçao do Hospital das Clinicas da Faculdade de Medicina da Universidade de Sao Paulo, Sao Paulo, Brazil - Dr. Ribeiro; Skejby Sygehus, Aarhus, Denmark - Dr. Thuesen; Rigshospitalet, Copenhagen, Denmark - Dr. Kelbaek; Cochin Hospital, Paris, France - Prof. Weber; Unité de Cardiologie Interventionnelle, Toulouse Cedex, France - Dr Fajadet; Institut Cardiovasculaire Paris Sud , Massy, France - Dr Morice; Hôpital Henri Mondor, Créteil, France - Prof. Teiger; Hôpital Rangueil, Toulouse Cedex, France - Dr Carrie; Herzkatheterlabor und Praxisklinik Andrea-Grüntzig-Haus, Hamburg, Germany - Prof. Dr med. Schofer; Medizinische Klinik 1 der RWTH Aachen, Germany - Prof. Hoffmann; Klinikum Villingen, Villingen, Germany - Prof. Jung; AK-St. Georg Hamburg, Hamburg, Germany - Prof. Kuck; Herz Zentrum- Bad Krozingen, Germany - Prof. Neumann; Segeberger Kliniken GmbH, Bad Segeberg, Germany - Prof. Richardt; Charité Universitätsmedizin Berlin, Germany - Prof. Stangl; Klinik für Kardiologie Westdeutsches Herzzentrum, Universitätsklinikum Essen, Germany, Dr Kahlert; Amphia Ziekenhuis, Breda, The Netherlands - Prof. Den Heijer; Catharina Hospital, Eindhoven, The Netherlands - Dr Koolen; University Medical Center Utrecht, The Netherlands - Dr Stella; St. Antonius Hospital, Nieuwegein, The Netherlands - Dr Suttorp; Mercy Hospital and North Shore Hospital, Auckland, New Zealand - Dr Ormiston; Christchurch Hospital, Christchurch, New Zealand - Dr McClean; Auckland City Hospital, Auckland, New Zealand - Dr Webster; Southampton Hospital, Southampton, UK - Dr Curzen; John Radcliffe Hospital, Oxford, UK - Dr Banning; Leeds Teaching Hospital NHS Trust, Leeds, UK - Dr Blackman; Brighton and Sussex University Hospital NHS Trust, Brighton, UK - Dr De Belder; Golden Jubilee Hospital, Clydebank, Scotland - Prof. Oldroyd; St. Thomas’ Hospital, London, UK - Dr Redwood
Funding
This trial is sponsored by Cordis, a Johnson & Johnson company.
Conflict of interest statement
A. Abizaid and J. Fajadet are members of Cordis scientific advisory boards and received minor honoraria. N. Macours and A. Qureshi are full-time employees of Cordis Corporation. C. Spaulding was a member of scientific advisory boards for Cordis and had received minor honoraria, and was a full-time employee of the Cordis Corporation between June 2010 and December 2011. The other authors have no conflicts of interest to declare.
