KEYWORDS
Abstract
Aims: Novolimus, a macrocyclic lactone with anti-proliferative properties, has a similar efficacy to currently available agents; however it requires a lower dose, and less polymer, and is therefore conceivably safer.
Methods and results: The EXCELLA II study was a prospective, multicentre, single-blind, non-inferiority clinical trial which randomised 210 patients with a maximum of two de novo coronary artery lesions in two different epicardial vessels in a ratio of 2:1 to treatment with either the Elixir DESyne Novolimus Eluting Coronary Stent System (NES n=139, Elixir Medical, Sunnyvale, CA, USA) or the Endeavor zotarolimus eluting stent (ZES n=71, Medtronic, Santa Rosa, CA, USA). The primary endpoint was in-stent mean late lumen loss (LLL) at 9-months follow-up. In-stent percent volume obstruction (%VO) was measured in a sub-group of 65 patients having 9-month intravascular ultrasound (IVUS) follow-up. Clinical secondary endpoints included a device orientated composite of cardiac death, target vessel myocardial infarction (MI), and clinically indicated target lesion revascularisation (CI-TLR) assessed at 9-months follow-up. At 9-months, the in-stent LLL was 0.11±0.32 mm in the NES arm, as compared to 0.63±0.42 mm in the ZES (p<0.0001 non-inferiority, p<0.0001 superiority). In-stent%VO was 4.5±5.1% and 20.9±11.3% for NES and ZES, respectively (p<0.001). There was no significant difference between stent groups in the device orientated composite endpoint (NES 2.9% vs. ZES 5.6%, –2.8% [-8.8%, 3.3%], p=0.45) or its individual components of cardiac death, target vessel MI and CI-TLR.
Conclusions: This non-inferiority randomised study not only met its primary endpoint, but also demonstrated superiority of NES compared to the ZES in terms of in-stent LLL.
Abbreviations
BMS: Bare metal stent
CABG: Coronary artery bypass graft
CI-TLR: Clinically indicated target lesion revascularisation
CK-MB: Creatinine kinase myoglobin fraction
DES: Drug eluting stents
DS: Diameter stenosis
EES: Everolimus eluting stent
IVUS: Intravascular ultrasound
LLL: Late lumen loss
MI: Myocardial infarction
MLD: Minimal lumen diameter
NES: Novolimus eluting stent
NQWMI: Non-Q-wave myocardial infarction
PCI: Percutaneous coronary intervention
PES: Paclitaxel eluting stent
QCA: Quantitative coronary angiography.
RVD: Reference vessel diameter
SES: Sirolimus eluting stent
TIMI: Thrombolysis in Myocardial Infarction
TVR: Target vessel revascularisation
ULN: Upper limit of normal
ZES: Zotarolimus eluting stent
Introduction
Drug eluting stents (DES) have revolutionised the field of percutaneous coronary intervention (PCI) since their inception in 2002.1 However, despite their undeniable efficacy at reducing neointimal proliferation and repeat revascularisation compared to their bare metal stent predecessors,2-5 they have been unable to completely eliminate restenosis, and in more recent times have been dogged by continued concerns over their long-term safety.6,7 These unresolved issues have prompted design modifications to historical DES systems, in an attempt to develop new DES systems which are both safer, and more efficacious.
The most widely used anti-proliferative agents on current DES systems such as sirolimus, zotarolimus and everolimus are all derived from macrocyclic lactones, and ultimately function through the inhibition of the mammalian target of rapamycin (mTOR), resulting in arrest of the cell cycle. Previous clinical studies have demonstrated the overall superiority, in terms of both reduced late lumen loss, and repeat revascularisation of coronary stents which elute mTOR inhibiting drugs when compared to those eluting paclitaxel.8
Novolimus is a metabolite of sirolimus that has been specifically developed for the Elixir DESyne Novolimus Eluting Coronary Stent System (NES, Elixir Medical, Sunnyvale, CA, USA). This modification was aimed at creating a new anti-proliferative drug, which had similar efficacy to currently available agents, but required a lower dose, and lower polymer load and therefore was conceivably safer. The feasibility of using novolimus on a DES has been assessed in the 15-patient first-in-man EXCELLA study, which reported an angiographic in-stent late loss of 0.31±0.25 mm, and a percent volume obstruction on intravascular ultrasound (IVUS) of 6.0±4.4% at 8-months follow-up, together with no major adverse cardiovascular events (MACE) through 12 months,9 and one MACE event at 24 months, which included a patient death wherein the patient had numerous other cardiovascular co-morbidities.10
Further assessment of the NES has been performed in the single-blind, prospective EXCELLA-II study, which randomised patients to treatment with either NES or the Endeavor (Medtronic, Santa Rosa, CA, USA) zotarolimus eluting stent (ZES). The current study reports the 9-month angiographic, IVUS and clinical outcomes of patients enrolled in the EXCELLA-II study, which represents the first, and largest randomised assessment of a coronary stent eluting novolimus.
Methods
Patient population
The EXCELLA-II study was a prospective, single blind, multi-centre trial enrolling 210 patients who were randomised in a ratio of 2:1 to receive either an Elixir NES (n=139), or an Endeavor ZES (n=71). All patients were over the age of 18, with evidence of myocardial ischaemia (as defined by the Canadian Cardiovascular society classification, or documented silent ischaemia or a positive functional test), and a maximum of two de novo native coronary artery lesions in different major epicardial vessels. For inclusion, on visual estimation, target lesion(s) were required to be: in a vessel with a reference vessel diameter between 2.5-3.5 mm; <24 mm in length; with a percentage diameter stenosis (DS) between 50-99%, and a Thrombolysis In Myocardial Infarction (TIMI) flow grade ≥1 by visual estimation.
Patients with documented evidence of recent (<3 days) myocardial infarction (creatinine kinase [CK]>2 times the upper normal limit [ULN]); a left ventricular ejection fraction <25%; a serum creatinine >2 mg/dl; those waiting heart transplantation; females of child-bearing age; those who would not consent to follow-up angiography; those with limited life expectancy due to concomitant disease; or those having a known sensitivity or contraindications to aspirin, ticlopidine, clopidogrel, mTOR inhibitor class drugs, cobalt chromium alloy, methacrylate or sensitivity to contrast which could not be adequately pre-medicated were excluded.
Angiographic lesions involving the left main stem; the aorto-ostial junction; those located within 5 mm of the origin of the left anterior descending or left circumflex artery; involving a side branch >2 mm in diameter; located within 10 mm of a previous stent; with heavy proximal calcification; requiring a staged procedure within 9-months; with a lesion with DS>40% proximal or distal to the target lesion; likely to require adjunctive therapy or which had associated visible thrombus were also all excluded. The ethics committee of each participating institution approved the study protocol, and all patients provided written informed consent.
Novolimus eluting stent
STENT PLATFORM
The Elixir DESyne Novolimus Eluting Coronary Stent System is comprised of the Elixir Core Coronary Stent System, which has received CE Mark approval in the European Union, and a novolimus eluting polymer coating. The Elixir core stent is a balloon expandable stent developed utilising a medical grade cobalt chromium alloy with a nominal strut thickness of 0.0032” (80 microns) including a 6-crown (2.5 mm stent diameter) and an 8-crown (3.0 and 3.5 mm stent diameter) two-link pattern designed to optimise vessel coverage, flexibility and deliverability.
POLYMER
The NES polymer is a durable poly n-butyl methacrylate (PBMA) polymer, which is similar to that currently in clinical use on medical devices including vascular implants and other DES systems such as the Cypher sirolimus eluting stent (SES, Cordis, Warren, NJ, USA) and the XIENCE V everolimus eluting stent (EES, Abbott Vascular, Santa Clara, CA, USA). Importantly the polymer undergoes substantial processing to purify it from unwanted impurities, thereby resulting in a reduction in its overall monomer content. The drug-polymer matrix is applied to the surface of the stent, without a primer polymer coating underneath, using a proprietary spray resulting in a coating thickness of <3 µm, which is thinner than that found on other currently available durable polymer DES (4.1 µm on ZES, 7.6 µm on EES). The polymer facilitates controlled release of novolimus, such that 80% of the drug is released over 12 weeks, with elution complete by 6-months (data on file at Elixir Medical).
NOVOLIMUS
Novolimus is a macrocyclic lactone which has been developed by removal of a methyl-group from carbon C16 (data on file at Elixir medical). Notably this differs from the other macrocyclic lactone agents that are used in DES, which have all been developed through modifications on the carbon C40 of the macrocyclic ring. Nevertheless, in a similar fashion to these other agents, novolimus binds to the immunophilin, FK Binding Protein-12 (FKBP-12), to generate an immunosuppressive complex, which binds to and inhibits the activation of the regulatory kinase mTOR. This inhibition suppresses cytokine-driven cell proliferation, inhibiting the progression from the G1 to the S phase of the cell cycle. Novolimus has been shown in in vitro studies to have a high potency to inhibit human smooth muscle cells (IC50 of 0.5 nM), which is comparable to that of sirolimus (data on file at Elixir Medical). The dose of Novolimus used on the DESyne stent is 5 µg/mm of stent length (compared to 10 µg/mm on ZES, and EES).
Study procedure
Patients were randomised by central telephone randomisation service in a ratio of 2:1 between NES and ZES after the identification of suitable lesions on preliminary angiography. Randomisation was stratified on a site level using random permuted block assignments. Physicians were not blinded, in view of the different packaging for each stent.
NES were available in diameters of 2.5, 3.0 and 3.5 mm and in lengths of 14, 18 and 28 mm, whilst ZES were available in diameters of 2.5, 3.0 and 3.5 mm and in lengths of 14, 18 and 30 mm.
Standard interventional techniques were used to treat the lesion, in particular pre-dilatation was mandatory, and stent implantation was performed at a pressure not exceeding the rated burst pressure. Each stent was required to be long enough to cover the lesion, and pre-dilated area including 2 mm on either side. Post-dilatation was left to the operator’s discretion; however, if performed, balloons were required to be shorter than the length of the deployed stent. In the event of a bailout procedure and the need for an additional stent, this was required to be of the same type as the first implanted stent if possible. If not, then a stent comprised on the same base material and drug family was recommended. In patients with two de novo lesions, attempts at the second lesion were only permitted if an optimal result defined as a residual DS<20%, TIMI 3 flow, absence of thrombus or edge dissection was seen after PCI of the first lesion.
In a subset of 65 (43 NES, 22 ZES) consecutive patients enrolled in pre-selected centres IVUS was performed after optimal stent placement had been achieved. Periprocedural pharmaceutical treatment was administered according to standard hospital practice. Procedural anticoagulation was achieved with unfractionated heparin or bivalirudin. The use of glycoprotein IIb/IIIa inhibitors was left to the operator’s discretion. All patients enrolled into the study were to receive ≥75 mg of aspirin daily for a minimum of one year, and clopidogrel 75 mg for a minimum of twelve months following the index procedure.
Follow-up
Patient clinical status review by telephone or hospital visit was planned at 1 (±14 days), 6, 9, and 12 months, and will be followed by annual review out to 5-years (±1 month). At outpatient visits, patients were specifically questioned about the development of angina or the occurrence of any adverse events. Angiographic follow-up for all patients was planned at 9-months (±1 month), with IVUS follow-up planned in a subset of 65 consecutive patients (from selected centres). Prior to follow-up angiography physicians were required to clinically evaluate the patients and prospectively record in the case record form whether any revascularisation, if required, was clinically indicated.
QCA
Quantitative coronary angiography (QCA) was performed using the CAAS II analysis system (Pie Medical BV, Maastricht, The Netherlands).11 In each patient, the stented segment and the peri-stent segments (defined as a length 5 mm proximal and distal to the stent edge) were analysed. The following QCA parameters were calculated: minimal lumen diameter (MLD), reference vessel diameter (RVD) obtained by an interpolated method, and DS%. Binary restenosis was defined in every segment as a DS≥50% at follow-up. Late lumen loss (LLL) was calculated as the difference between the post-procedure and follow-up MLD. Angiography films were centrally assessed at one angiographic core laboratory (Cardialysis, Rotterdam, The Netherlands) with assessors unaware of the allocated stent.
IVUS
IVUS assessments were performed immediately post-stent implantation and at the 9-month follow-up time frame in a subset of 65 patients. Standard IVUS procedures were followed including a motorised pullback at 0.5 mm/sec from the distal reference segment, to at least 10 mm proximal to the lesion/stent border. Quantitative IVUS parameters included the minimum lumen area, and volume data for the neointimal hyperplasia, vessel, stent, and lumen. To standardise for different lengths, the volume index was calculated as volume divided by length. The Cardiovascular Core Analysis Laboratory (CCAL) at Stanford University Medical Centre, Stanford, CA, USA performed the independent analysis of the lesions.
Study endpoints
The primary endpoint of the study was in-stent LLL as assessed by QCA at 9-months follow-up. Secondary QCA/IVUS endpoints included: MLD and DS% post-procedure and at 9-months; 9-month in-segment LLL; 9-month in-stent and in-segment binary angiographic restenosis DS≥50%; and in the subset having 9-month IVUS follow-up, in-stent percentage volume obstruction (%VO). In-stent was defined within the margins of the stent, while in-segment was defined as located within the margins of the stent and 5 mm proximal or distal to the stent.
Secondary clinical endpoints, collected at 1, 6, and 9 months, and will be collected annually to 5-years included the device-orientated composite endpoint defined as cardiac death, MI not clearly attributable to a non-intervened vessel (World Health Organisation [WHO] definition), and clinically indicated target-lesion revascularisation (CI-TLR) by PCI or bypass surgery. Other secondary clinical endpoints were CI-TLR, clinically indicated target vessel revascularisation (CI-TVR), stent thrombosis defined according to the Academic Research Consortium (ARC) definition,12 and acute success including both clinical device and clinical procedural success.
An independent blinded clinical events committee (CEC) evaluated all clinical endpoints, and a Data and Safety Monitoring Board, neither of which were affiliated with the study, ensured the safe conduct of the trial.
Definitions
All deaths were considered cardiac unless an undisputed non-cardiac cause was present. MI was defined according to the WHO definition13 wherein Q-wave MI was defined as the development of new pathological Q-waves in association with a rise in CK ≥2 times the ULN. A non-Q-wave MI (NQWMI) was defined as a CK elevation ≥2 times the ULN together with an elevation in CK-MB. A target lesion revascularisation was considered clinically indicated if angiography at follow-up showed a DS<50% (core lab QCA assessment) and if one of the following occurred: (1) a positive history of recurrent angina pectoris, presumably related to the target vessel; (2) objective signs of ischaemia at rest (ECG changes) or during exercise test (or equivalent), presumably related to the target vessel; (3) abnormal results of any invasive functional diagnostic test (e.g., fractional flow reserve); (4) A TLR with a diameter stenosis ≥70% even in the absence of the above-mentioned ischaemic signs or symptoms. Device success was defined as the attainment at the target site of a final residual DS<20% using only a NES or ZES stent alone. Procedure success was defined as the attainment at the target site of a final residual DS<50% using a NES or ZES stent alone, together with the absence of any in-hospital device orientated composite endpoints.
Statistical methods
The sample size for this study was calculated based on the planned analysis of the primary endpoint using a one-tailed t-test to show non-inferiority of the test arm compared to the control arm at the 0.05 significance level. Assuming the in-stent late lumen loss at nine months was equivalent between the test and control, using a common standard deviation of 0.5 and a margin of equivalence (delta) of 0.20, in order to have an 80% probability (i.e., 0.80 power) a minimum of 118 test and 59 control patients were required for the study. To allow for an approximate 15% patient dropout/lost-to-follow-up rate, approximately 210 patients of which 139 were test and 71 control patients were enrolled.
Binary variables are presented as percentages, and compared using the Fisher’s exact test. Continuous variables were compared using the t-test, apart from the non-inferiority primary endpoint of in-stent LLL which was analysed using SAS v8 Proc Mixed which took into account the within-patient correlation structure of these data. The hypothesis testing for the primary endpoint was performed using a one-sided non-inferiority test with asymptomatic test statistic. If non-inferiority was shown, superiority analysis was planned using a two-sided t-test at the 5% alpha level. Clinical outcomes were analysed on an intention to treat basis using all patients randomised in the study, regardless of the treatment actually received. QCA data was analysed on a modified intention to treat basis, where patients were only included if at least one study stent was implanted, and lesions were only included if they were treated with a study stent. Statistical analyses were performed using the SAS statistical package (version 9.1.3, SAS Institute, Cary, NC, USA).
Results
The EXCELLA II study screened 622 patients over 21 sites, of whom 210 (33.8%) were randomised to treatment with NES (n=139) and ZES (n=71) between 28 October 2008 and 5 March 2009. At 9-month follow-up clinical assessment, as shown in Figure 1, was available in 207 patients (98.6%), made up of 137 of the initial 139 NES patients (98.6%) and 70 of the original 71 ZES patients (98.6%). Of the two patients failing to complete 9-month follow-up in the NES group, one was lost to follow-up after 6-months, whilst one withdrew consent prior to the 9-month follow-up angiogram. A single patient in the ZES was lost to follow-up after 4-weeks. Importantly no patient experienced any events up to last patient contact. Overall angiographic follow-up on a modified intention to treat basis was available in 88.6% of patients (89.9% NES, 85.9% ZES), with the reasons for incomplete follow-up shown in Figure 1.
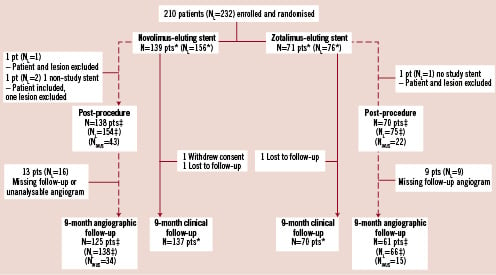
Figure 1. Clinical and angiographic follow up of patient population. Pts: patients; NL: number of lesions; IVUS: intra-vascular ultrasound; *Intention to treat analysis; ‡ Modified intention to treat: lesions with at least 1 study stent
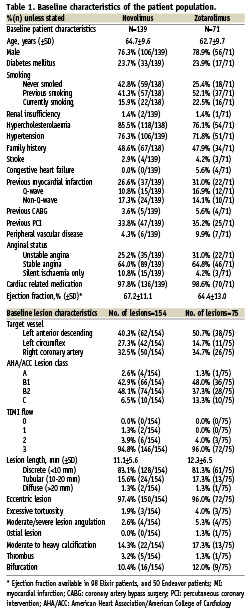
Patient population and lesion characteristics
Baseline patient and angiographic characteristics are shown in Table 1. Overall, both groups had similar baseline risk profiles, and comparable lesion characteristics. Clinical device success occurred in the 137 out of 156 lesions in the NES arm (87.8%), and 70 out of 76 lesions in the ZES arm (92.1%). Of the 19 NES device failures, four were the result of implantation of a non-study stent, whilst 15 occurred because QCA deemed the residual DS to be >20% (mean±SD: 22.3±2.0%). Of note in 12 of these patients the investigator reported the residual DS to be 0%. Similarly in the ZES arm, one non-study stent was implanted, whilst five lesions had a residual DS on QCA of >20% (22.4±1.9%); all of these had been deemed to have a DS of 0% by the investigator. The rates of procedural success were NES 97.8%, and ZES 98.6%, (–0.7% [–4.4%, 2.9%], p=1.00).
QCA analysis
Angiographic data at 9-months were available in 186 patients (204 lesions), and are summarised in Table 2. The RVD, MLD and DS% pre- and post procedure were all comparable between both study arms. At 9-months follow-up however, there was a larger RVD, a significantly larger MLD, and both a significantly lower DS% and rate of in-stent binary restenosis following treatment with NES. At 9-months, the primary endpoint of mean in-stent LLL was significantly lower for NES compared to the ZES, (0.11±0.32 mm vs. 0.63±0.42 mm, non-inferiority p<0.0001, superiority p<0.0001). The cumulative frequency distribution of in-stent LLL and DS% is displayed in Figure 2.
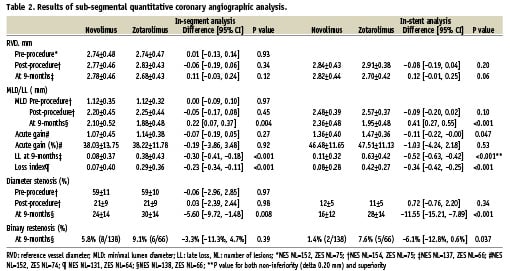
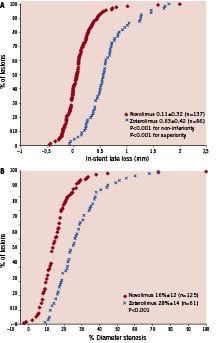
Figure 2. Cumulative frequency of (A) in-stent late loss, (B) % diameter stenosis at 9-month follow-up.
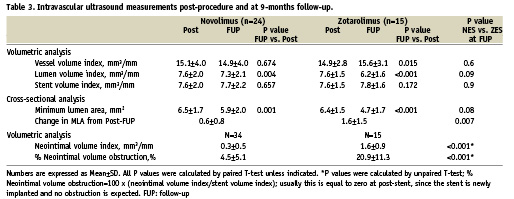
IVUS analysis
Baseline and follow-up IVUS data, which was obtained in 50 patients, including 39 serial sets of data (24 NES, 15 ZES) are shown in Table 3. At follow-up, there were no significant differences between NES and ZES with respect to the absolute vessel, lumen or stent volume indexes. Volumetric analysis indicated significantly less neointimal hyperplasia, (0.3±0.5 vs. 1.6±0.9, p<0.001) and %volume obstruction (4.5±5.1 vs. 20.9±11.3, p<0.001) with NES compared to ZES.
Incomplete stent apposition post procedure was evident in 9/41 (22%) NES lesions, three of which were post-dilated; and 2/23 (8.7%) ZES lesions, both of which were post-dilated. Serial analysis indicated that incomplete stent apposition at 9-months was persistent or resolved in 3/31 (9.7%) and 2/31 (6.5%) NES lesions, and 1/19 (5.3%) and 1/19 (5.3%) ZES lesions, respectively. There were no cases of late acquired incomplete stent apposition.
Clinical outcomes
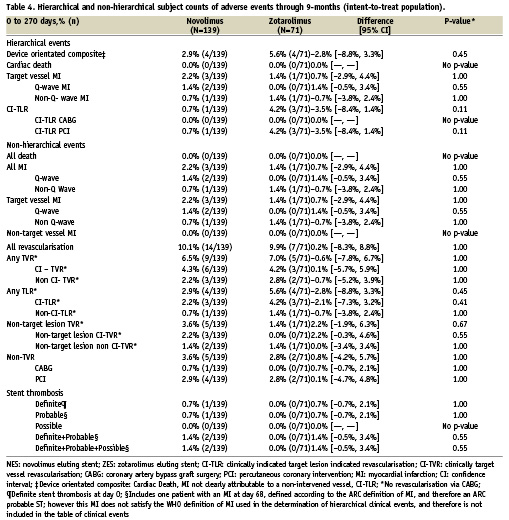
Hierarchical and non-hierarchical clinical outcomes are shown in Table 4. Overall, there was no significant difference between stent groups in the device orientated composite endpoint (NES 2.9% vs. PES 5.6%, –2.8% [–8.8%, 3.3%], p=0.45) or its individual components of cardiac death, target vessel MI and CI-TLR. In total four MIs (two Q-wave and two NQWMI) were recorded in the study population, all of which were attributable to the study vessel. All TVR, which was performed in 6.5% and 7.0% of patients receiving NES and ZES respectively, was performed with PCI.
Specific to the Q-wave MI patients, one patient experienced a spiral dissection of the LAD during implantation of the first stent, and in the process of treating the dissection the operator implanted three more study stents. At the end of the procedure there was TIMI 2 flow in the target vessel, but within 4-hours, repeat angiography performed due to chest pain revealed an occlusion of the LAD, which required additional treatment with thrombus aspiration and balloon dilatation. The distal segment of the LAD remained occluded. This patient was adjudicated by the CEC as having had a Q-wave MI, a TLR due to DS100%, and a definite acute stent thrombosis. The other Q-wave MI was decided by default, after the CEC could not decide whether the patient had experienced a Q-wave or NQWMI. In this case the procedure itself went without incident, however a CK rise of 614, and CK-MB of 48.3 was recorded post intervention. In the two NQWMI patients, one had an unremarkable procedure, whilst the other had clear evidence of distal embolisation.
Stent thrombosis rates, as shown in Table 4, were low, with only two ST events in patients treated with NES (one early definite, and one late probable), and no ST observed with ZES. The definite ST event was an acute ST, occurring within four hours of the index PCI procedure, which was complicated by a spiral dissection necessitating the implantation of three additional study stents as described earlier. The ST in this case was attributed to procedural complications. The probable ST occurred in a patient still on dual antiplatelet therapy, who was admitted with dyspnoea and no chest pain, however the patient was found to have an elevated troponin, and had ischaemic ECG changes in the territory of the stent implanted at the index PCI 68 days earlier.
Discussion
The main findings from this study are that a specifically designed DES eluting novolimus has a significantly lower 9-month in-stent LLL compared to a ZES, with comparable clinical outcomes at 9-months.
Novolimus represents a specifically manufactured macrocyclic lactone, which is a metabolite of sirolimus that advantageously results in lower concentrations of drug (and consequently polymer) being needed to inhibit neointimal proliferation. The dose of drug and polymer thickness on NES are 5 µg/mm and <3 µm respectively, compared to 10 µg/mm and 4.1 µm for ZES; 10 µg/mm and 7.6 µm for the EES; 10 µg/mm and 16 µm for the TAXUS paclitaxel eluting stent (PES, Boston Scientific, Natick, MA, USA); and 14 µg/mm and 12.6 µm for SES. Although the NES utilises a methacrylate polymer that is similar to that used in other commercially available DES, the absence of a primer coating, together with purification of the polymer, and lower doses of drug and polymer conceivably have advantages when considering that a hypersensitivity reaction to the presence of a polymer is thought, in part, to be responsible for precipitating ST.14-18
There are two notable differences between the performances of NES in the current study, and the previously reported FIM. Firstly, there is a marked reduction in the late loss from the 0.31±0.25 mm reported in the FIM study to the 0.11±0.32 mm seen in this study. These results, in the absence of any changes in stent specification, may relate to the small sample size in the FIM study. In addition late loss is widely recognised to have a rather large standard deviation when inter-observer variability is assessed; moreover the measure itself is made up of two individual measurements, each with their own inter-observer variability, which is due mainly to the process of calibration.11,19 Finally both studies were performed in different angiographic core labs, using different analysis equipment possibly leading to inter-lab variability. The second notable difference between both studies is the rate of incomplete stent malapposition amongst lesions in the IVUS sub-group which was 43% in the FIM study and 22% in the current study. In the absence of any changes to the stent’s radial strength, or any changes to the protocol for post-stent dilatation between the two studies, it is again likely that these differences are related to the small sample size of the FIM study, together with inter-observer variability.
Two important topical issues with respect to the evaluation of new DESs, namely the definition of clinical endpoints such as MI and the selection of an appropriate control stent, are highlighted by the present study. Firstly, the current report defines MI according to the historical definition from the WHO, which was primarily selected to enable comparability of the current results with those of previous DES studies. It is noteworthy that despite being published in 2007 the ARC and the Universal definitions for MI12,20 are still not utilised in recently published pivotal trials of first and second generation DESs such as Endeavor III (ZES vs. SES), Endeavor IV (ZES vs. PES) and COMPARE (EES vs. PES).21-23 In fact, the RESOLUTE all-comers trial, which is due for publication later this year, will be the first study to report MI according to both historical and ARC definitions.24 Conversely, interventional studies investigating new anti-coagulant agents such as ticagrelor have been quick to utilise the new Universal definitions.25 This variation between device and pharmacological studies suggests that a definition of this important clinical endpoint, that is satisfactory to all parties, is yet to be established.
The selection of an appropriate control stent in the assessment of a new DES is an important aspect of trial design. Some may question the use of ZES as the control stent in the current study, when at present the market leader is considered to be the EES. Of note, authorities do not accept first-in-man studies for regulatory approval, instead they require randomised studies against a comparator DES, however this is not required to be the most efficacious or safest stent at the time of study. This subsequently allows non-inferiority trials to be designed without prohibitively large sample sizes.
At the time of the study design in 2007, in the aftermath of the “Barcelona firestorm” where first generation DES were reported as causing an increased risk of very late ST,7,26,27 ZES was considered the safest DES, although contemporary data now indicate that this may not now be the case.28 Meta-analysis have confirmed a 1.4-3.5 times increased risk of very late ST with SES and PES compared to BMS out to 4-year follow-up.6 On the other hand, in the randomised ENDEAVOR II study the rate of very late ST at 5-years follow-up was only 0.2% with ZES, compared to 0.3% for the BMS controls.29 These differences in very late ST rates prompted the recently enrolled 8,800 patient PROTECT study which will be the only study adequately powered to report differences in ST between ZES and SES.30
We acknowledge that with respect to inhibition of neointimal proliferation, when ZES has been compared to SES and PES in the respective ENDEAVOR III, and ENDEAVOR IV studies, a significantly higher LLL of 0.60 mm and 0.67 mm respectively has been seen with ZES at 8-months follow-up. These values however are comparable to the 0.33 mm LLL seen with the NES in the Excella FIM study, thereby allowing a feasible non-inferiority study between NES and ZES to be performed.9,22,31 Importantly, despite this higher LLL, no significant differences in TLR have been seen with ZES at 36- or 48-months compared to PES and SES, respectively.29,32 Moreover, reductions in the absolute difference in TLR between short and long-term follow-up in favour of ZES indicate that ZES may not be susceptible to the delayed restenosis phenomenon observed with other mTOR inhibiting DES such as SES,33 and EES.34
Limitations
This study is limited by its short duration of follow-up, lack of blinding of the operator, and lack of power to discriminate between clinical events.
Conclusions
This non-inferiority randomised study not only met its primary endpoint, but also demonstrated superiority of the novolimus eluting stent compared to the zotarolimus eluting stent in terms of in-stent late loss. In addition, rates of adverse cardiac events were low and comparable between both stents.
Appendix
Sponsor: Elixir Medical Corporation, Sunnyvale, CA, USA
Principal investigator: Patrick W. Serruys, Erasmus Medisch Centrum, Rotterdam, The Netherlands
Co-principal investigators: Stephan Windecker, University Hospital Bern, Bern, Switzerland; John Ormiston, Auckland City Hospital and Mercy Angiography, Auckland, New Zealand; Alexandre Abizaid, Instituto Dante Pazzanese, Sao Paulo, Brazil
Steering committee: Patrick W. Serruys, Principal Investigator and Chairman, Rotterdam, The Netherlands; Stephan Windecker, Bern Switzerland; John Ormiston, Auckland, New Zealand; Alexandre Abizaid, Sao Paulo, Brazil; Peter Fitzgerald, Cardiovascular Core Analysis Laboratory, Stanford University, Stanford, CA, USA; Elixir Medical Corporation, Sunnyvale, CA, USA
Data Safety Monitoring Board (DSMB): Prof. Dr. J.G.P. Tijssen, Chairman, Amsterdam, The Netherlands; Dr. B.J.W.M. Rensing, Nieuwegein, The Netherlands; Dr. M.J.B.M. van den Brand, Ouderkerk aan den Ijssel, The Netherlands
Clinical Events Committee (CEC): Dr. Pascal Vranckx, Luik, Belgium; Dr. Peter Smits, Rotterdam, The Netherlands; Dr. Anthony H. Gershlick, Glenfield, United Kingdom; Prof. Victor Legrand, Liège, Belgium
Data Management - Angiographic Core Laboratories: Cardialysis BV, Rotterdam, The Netherlands
Data Management - IVUS Core Laboratories: Cardiovascular Core Analysis Laboratory, Stanford University, Stanford, CA, USA
Data Coordination Centre and Site Monitoring: Cardialysis BV, Rotterdam, The Netherlands, Krakow Cardiovascular Research Institute (KCRI), Krakow, Poland, Medical Device Consultancy, Christchurch, New Zealand, Pacific Clinical Research Group (PCRG), Mosman, NSW, Australia, Kerstin Kupfer, Australasian Ltd, Bonn, Germany, Anton Maierl, München, Germany
The following investigators and institutions participated in the EXCELLA II Trial:
Patrick W. Serruys, Prof. MD, Erasmus Medisch Centrum, Rotterdam, The Netherlands; John Ormiston, MD, Auckland City Hospital and Mercy Angiography, Auckland, New Zealand; Stefan Verheye, MD, AZ Middelheim Hospital, Antwerp, Belgium; Christophe Dubois, MD, Universitair Ziekenhuis Gasthuisberg, Leuven, Belgium; Karl E. Hauptmann, MD, Krankenhaus der Barmherzigen Brüder, Trier, Germany; Jim Stewart, MD, Mercy Angiography, Auckland, New Zealand; Marcus Wiemer, MD, Herz-und Diabetes Zentrum, Bad Oeynhausen, Germany; Joachim Schofer, Prof. MD, Universitäres Herz- und Gefäßzentrum, Hamburg, Germany; Wolfgang Rutsch, Prof. MD, Charite Campus Charité Mitte, Berlin, Germany; Karl Stangl, Prof. MD, Charite Campus Charité Mitte, Berlin, Germany; Bernhard Witzenbichler, Prof. MD, Charite Benjamin Franklin Campus, Berlin, Germany; Emanuelle Barbato, MD, OLV Hospital Cardiovascular Center, Aalst, Belgium; Mark Webster, MD, Auckland City Hospital, Auckland New Zealand; Dariusz Dudek MD, PhD, Jagiellonian University, Krakow, Poland; Seif El-Jack, MD, North Shore Hospital, Auckland, New Zealand; Dougal McClean, MD, Christchurch Hospital, Christchurch, New Zealand; Patrick Kay, MD, Middlemore Hospital, Auckland, New Zealand; Sabine Genth-Zotz, MD, Klinikum der Johannes Gutenburg-Universität, Mainz, Germany; Pawel Buszman, Prof. MD, Upper-Silesian Heart Centre, Katowice, Poland; Ian Meredith, Prof. MD, Monash Medical Center, Melbourne, VIC, Australia; Thomas Ischinger, Prof. MD, Klinikum Bogenhausen der Stad Muenchen, Munich, Germany; Yves Taeymans, Prof. MD, University Hospital Gent, Gent, Belgium; Robert Whitbourn, Prof. MD, St. Vincent’s Hospital, Melbourne, VIC, Australia
