Abstract
Aims: To compare the efficacy and safety of the MiStent absorbable polymer sirolimus-eluting stent (APSES) with a zotarolimus-eluting stent (ZES).
Methods and results: The trial was a 2:1 randomisation at 26 sites of 184 patients implanted with an APSES (n=123) versus a ZES (n=61). Following stent implantation, all patients underwent quantitative coronary angiography at baseline and at nine months of follow-up, while a select subgroup also underwent optical coherence tomography (OCT). The primary efficacy hypothesis was superiority of in-stent late lumen loss (LLL) of APSES compared to ZES. At nine months, the primary endpoint was met, with a mean in-stent LLL of 0.27±0.46 mm in 103 APSES patients versus 0.58±0.41 mm in 52 ZES patients (p<0.001). The proportion of uncovered stent struts by OCT at nine months was very low in both groups. The mean neointimal thickness of covered struts (p=0.002) and percent net volume obstruction (p≤0.003) were significantly lower in the APSES than in the ZES group. Major adverse cardiac event and stent thrombosis rates were low and comparable between groups.
Conclusions: The DESSOLVE II trial demonstrated superiority in the primary efficacy endpoint of nine-month mean LLL for APSES compared to ZES. Strut coverage by OCT was high with both stents and the clinical safety endpoints including stent thrombosis were equally low in both groups. ClinicalTrials.gov Identifier: NCT01294748
Introduction
Drug-eluting stents (DES) were designed to prevent the development of coronary restenosis caused by the proliferation of neointimal tissue following stented angioplasty. The first generation of DES consisted of bare metal stents (BMS) coated with a permanent polymer, which eluted a drug that inhibited the growth of excessive luminal tissue. However, the presence of these durable polymers may contribute to the undesirable long-term effects on vessel healing potentially associated with (very) late stent thrombosis (ST)1,2.
In an effort to enhance safety and efficacy, current DES focus has shifted to developing strategies to eliminate the potential harm associated with permanent polymer-based DES. One of these solutions is the use of bioabsorbable polymers. The MiStent® Sirolimus Eluting Absorbable Polymer Coronary Stent System (MiStent SES®; Micell Technologies, Durham, NC, USA) was developed using a thin-strut cobalt-chromium (CoCr) stent platform with a fully absorbable polymer coating uniquely containing a crystalline form of sirolimus. This next-generation sirolimus-eluting stent (SES) elutes and delivers sirolimus at a controlled rate from a recognised absorbable polymer (AP) and is intended to combine the long-term safety and stability characteristics of a BMS with the demonstrated clinical advantages of a DES.
We present the angiographic and the optical coherence tomography (OCT) evaluations along with clinical outcomes at nine months.
Study design and objectives
DESSOLVE II was a prospective, single-blind, 2:1 unbalanced, randomised, multicentre, superiority trial designed to compare the MiStent absorbable polymer sirolimus-eluting stent (APSES) to the control Endeavor® zotarolimus-eluting stent (ZES) system (Medtronic Vascular, Inc., Santa Rosa, CA, USA) using in-stent late lumen loss (LLL) at nine months as the primary study efficacy endpoint3. Investigational sites in Europe and New Zealand (Online Appendix A) participated in the trial.
Following implant of the DES, all patients underwent quantitative coronary angiography (QCA) and a subgroup underwent OCT at baseline and nine-month follow-up.
Patient population and methods
Patients included in this trial were required to be between the ages of 18 and 85 years, who presented with: a) stable or class I-IV unstable angina pectoris, or documented overt or silent myocardial ischaemia, and b) a single, de novo, type A, B1 or B2, >50% coronary stenosis (by visual estimate) in a 2.5 to 3.5 mm diameter (by visual estimate) native coronary artery that could be covered with a ≤30 mm long stent. Total occlusions, in-stent restenosis, highly calcified or thrombotic lesions and lesions located at major bifurcations or in highly tortuous vessels were excluded from the study. Direct stenting was not allowed. Patients could be enrolled ≥72 hrs after the onset of an acute myocardial infarction (MI), if the serum cardiac enzyme concentrations had returned to within normal ranges. All patients granted informed consent to participate in the study, which was approved by the ethics committees of the enrolling medical centres. The main study exclusion criteria are listed in Online Appendix B.
Study endpoints
PRIMARY
The primary efficacy endpoint of the study was in-stent LLL at nine-month follow-up by an independent angiographic core laboratory (Online Appendix A) , defined as the difference between the post-procedural minimal luminal diameter (MLD) and the follow-up MLD. The safety endpoint was the rate of major adverse cardiac events (MACE), defined as all-cause death, Q-wave or non-Q-wave MI and clinically driven target vessel revascularisation (TVR) at nine-month follow-up.
SECONDARY
The secondary procedural/angiographic endpoints of the study were device, lesion and procedural success, in-segment LLL, percent diameter stenosis (%DS), MLD, and binary restenosis rates at nine-month follow-up. The secondary clinical endpoints of the study ascertained immediately after the index procedure and at each subsequent follow-up were all-cause mortality, rates of Q-wave and non-Q-wave MI, rates of clinically driven target lesion revascularisation (TLR) and TVR, rates of target lesion failure (TLF) and target vessel failure (TVF), and rates of ST according to the Academic Research Consortium (ARC) definitions4 and adjudicated by an independent clinical events committee.
Device characteristics
APSES
The design of the rapid exchange delivery catheter of the MiStent APSES is compatible with guidewires ≤0.014 inch in diameter and 6 Fr guide catheters. The BMS platform of the APSES is a thin, laser-cut and electro-polished L605 cobalt-chromium alloy tube. The <10 µm thick (biased to the abluminal surface) stent coating consists of poly-lactide-co-glycolic acid (PLGA) loaded with crystalline particles of sirolimus (Figure 1)5. The drug load on the MiStent SES is similar to the CYPHER stent (Cordis, Miami Lakes, FL, USA) with a nominal sirolimus drug density of 2.44 µg/mm2. PLGA, a biodegradable and biocompatible polymer, releases the crystalline drug in a controlled manner post implant and throughout the stented region. Based on observations made in animals, all of the polymer coating of the APSES is expected to be off the stent in 45-60 days, leaving a BMS within the artery. The drug continues to elute from the crystals into the tissue up to nine months.

Figure 1. MiStent SES utilises crystalline sirolimus in a polymer coating (scanning electron microscopy).
ZES
The control Endeavor ZES combines the Driver® chromium-cobalt-nickel alloy coronary stent system with 91 µm stent struts coated with a durable phosphorylcholine polymer and 10 µg per 1 mm stent length of the antiproliferative agent zotarolimus6,7.
Antithrombotic therapy
All patients were treated with DAPT for a minimum duration concordant with local practice guidelines. A loading dose of aspirin was recommended before the procedure, followed by an oral maintenance dose daily, indefinitely. A loading dose of clopidogrel was recommended within 24 hrs before the procedure, followed by a maintenance dose for 6-12 months. Prasugrel could also be used for patients who were intolerant of clopidogrel.
Patient follow-up
Between the index DES implantation procedure and their return at nine months to undergo follow-up QCA, the patients were contacted at 30 days and six months to review their medication regimen and ascertain their clinical status and the possible incidence of interim adverse events. The patients will be contacted annually through five years.
Follow-up imaging at nine months
All available patients were seen at nine months for recording of an ECG, assessment of clinical status, drug regimen and possible interim adverse events, and repeat invasive angiography for QCA of the stented lesion by independent core laboratory analysis. OCT studies were conducted in a subgroup of 38 patients (24 APSES and 14 ZES). Sequential measurements were made during the index procedure, after DES deployment, and at nine months after administration of intracoronary nitrates. OCT images were acquired with a commercially available system (C7-XR™ OCT Intravascular Imaging System; St. Jude Medical, St. Paul, MN, USA) without proximal balloon occlusion after intracoronary administration of 200 µg of nitroglycerine through conventional guiding catheters. A 0.014 inch guidewire was positioned distally and the OCT catheter (C7 Dragonfly™; St. Jude Medical) was advanced distal to the region of interest, which was scanned using the integrated automated pullback device at 20 mm/s and acquiring images at 100 frames/s. During image acquisition, coronary blood flow was replaced by contrast dye infusion delivered with an injection pump at 3-5 ml/s via the guiding catheter.
Study monitoring
All sites and data were monitored during the trial and the co-principal investigators reviewed the final trial results. A clinical events committee met regularly to adjudicate pre-specified events including all deaths, MI, TVR and ST. The data safety monitoring board was responsible for the overall safety of the study.
Core laboratory analysis
QCA was performed as previously described8 using validated software (Medis version 7.2; Medis, Leiden, The Netherlands). Measurements included LLL, the primary study endpoint, as well as %DS, MLD, and binary restenosis (reduction in the %DS of 50% or more) of the treated lesion and vessel.
OCT measurements were obtained with a dedicated semi-automated contour-detection system (OCT system software B.0.1; St. Jude Medical). All cross-sectional images (frames) were initially screened for quality assessment and excluded from analysis if any portion of the image was out of the screen or the image had poor quality caused by residual blood or sew-up artefact9. Side branches occupying >45° of the cross-section were excluded from the analysis. Strut-level analysis was performed considering every three frames (0.6 mm interval) along the entire target segment. Lumen, stent, and neointimal hyperplasia areas and volumes were calculated9-11. The centre of the luminal surface of the strut blooming was determined for each strut and its distance to the lumen contour was calculated automatically to determine strut-level intimal thickness. Strut malapposition was determined when the negative value of strut-level intimal thickness was higher than the strut thickness, according to the stent manufacturer’s specifications, with the addition of a compensation factor of 20 µm to correct for strut blooming12. The blooming compensation factor was determined based on analysis of 2,250 struts. Highly reproducible measurements for strut apposition and coverage using the described methodology have been reported13. The following OCT variables were collected: percentage of stent struts uncovered/malapposed, percentage of frames with more than 30% of uncovered struts, maximum length of segments with uncovered/malapposed struts, maximum malapposition distance, percent net volume obstruction, frequency of abnormal intra-stent tissue, stent volume, and lumen volume. Qualitative imaging assessment was performed in every frame for the presence of abnormal intra-stent tissue which was defined as any mass protruding beyond the stent struts into the lumen, with irregular surface and a sharp intensity gap between mass and neointimal tissue14,15.
Statistical analysis
The primary efficacy hypothesis of the trial was superiority of in-stent LLL associated with APSES compared to ZES at nine months. A total of 184 patients were randomised in a 2:1 manner to APSES versus ZES. This sample size provided 90% power to demonstrate a >50% reduction in LLL with APSES with a two-sided alpha of 0.05, assuming a control LLL for ZES of 0.50 mm±0.48 mm and 20% loss to follow-up. For safety, the difference in nine-month rates of MACE between the APSES and ZES arms was estimated using a difference in sample proportions with a corresponding 95% confidence interval. Continuous variables were expressed as mean ±SD and categorical variables were expressed as counts and percentages. For per-patient analysis, the difference between the two stent types was evaluated by non-parametric Mann-Whitney U test for optical coherence tomography measurements, and Student’s t-test for quantitative coronary angiographic continuous variables and Fisher’s exact test for categorical variables. All statistical analyses were performed by Harvard Clinical Research Institute, Boston, MA, USA, using the Statistical Analysis Software, version 9.1.3 SP2 (SAS Corporation, Cary, NC, USA). Statistical significance was declared when the two-sided p-value was <0.05.
Results
Between February and July 2011 a total of 184 patients were randomised in DESSOLVE II: 123 were assigned to receive APSES and 61 to receive ZES. The mean age of the APSES patients (69% men) was 65.0±10.4 and that of the ZES patients (74% men) was 65.1±10.5 years. The baseline characteristics of the two study groups are shown in Table 1. Three patients did not correctly receive the assigned stent, including two patients assigned to APSES and one patient assigned to ZES, leaving 181 patients (121 APSES, 60 ZES) treated per protocol (Figure 2). However, the analysis is reported for the intention-to-treat group.
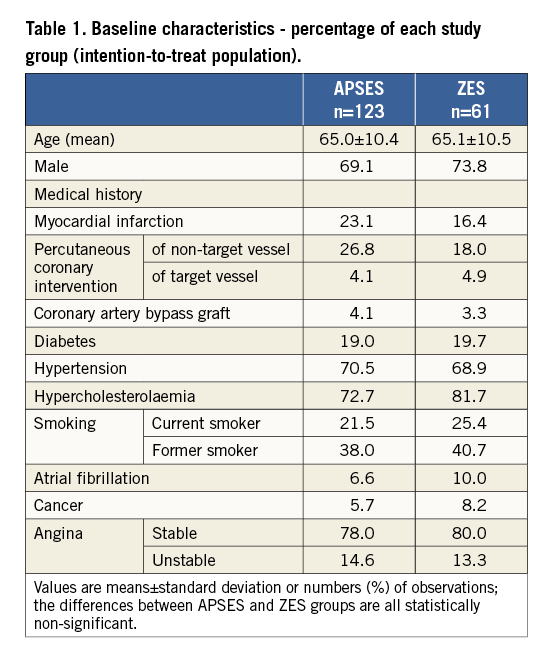
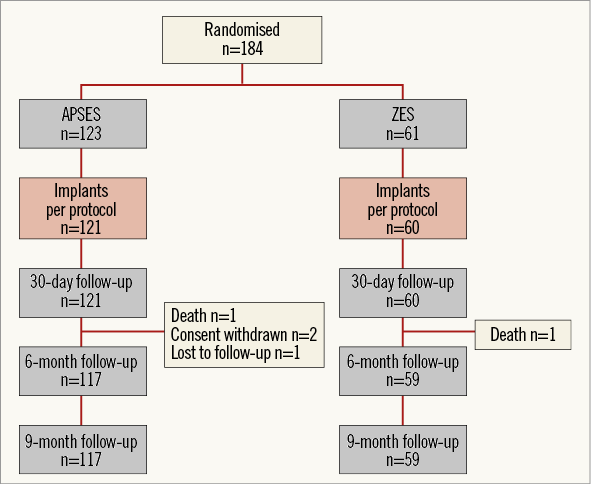
Figure 2. Patient randomisation and follow-up through 9 months.
Procedural and lesion characteristics
The angiographic lesion and procedural characteristics are shown in Table 2. Except for a minimally greater mean post-procedural %DS in the APSES group favouring the ZES group, all between-group differences were well balanced, including vascular lesion distribution, lesion length, amount of calcification, lesion complexity and mean post-procedural MLD.
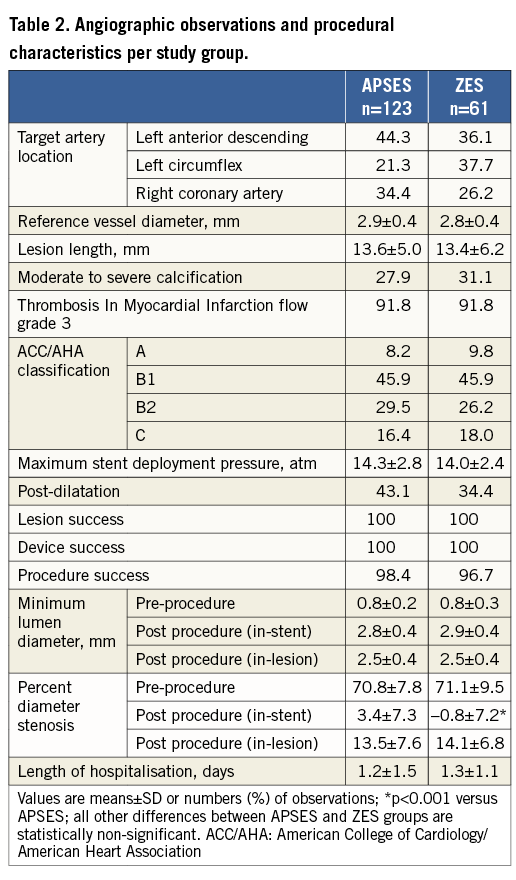
Quantitative coronary angiography and primary study endpoint analysis
The nine-month mean in-stent LLL was 0.27±0.46 mm in the APSES arm (n=103), versus 0.58±0.41 mm in the ZES arm (n=52) (Table 3). This -0.31 mm difference (95% CI: -0.45 to -0.16) between the two groups was statistically significant (p<0.001) in favour of APSES, confirming the primary efficacy hypothesis of the trial (Figure 3). Concordantly, mean in-stent and in-lesion MLD was significantly smaller, and mean in-stent and in-lesion %DS significantly greater in the ZES than in the APSES group (Table 3). Binary restenosis was similar in both groups.
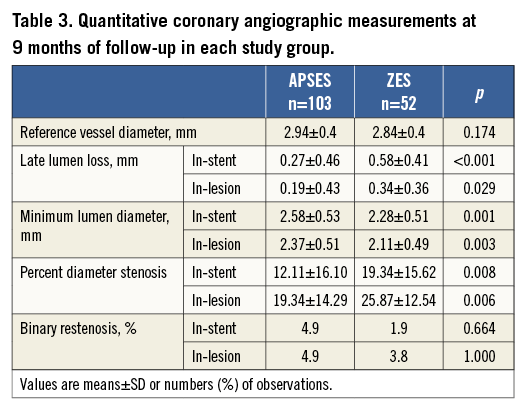
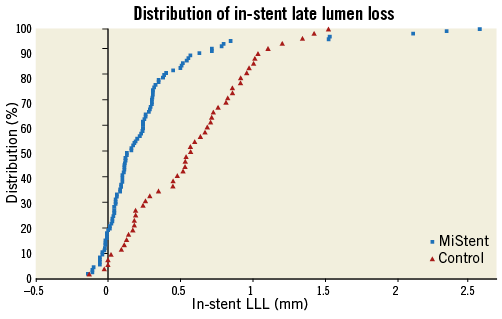
Figure 3. Cumulative distribution curves of the in-stent late lumen loss between groups (primary study endpoint).
Optical coherence tomography
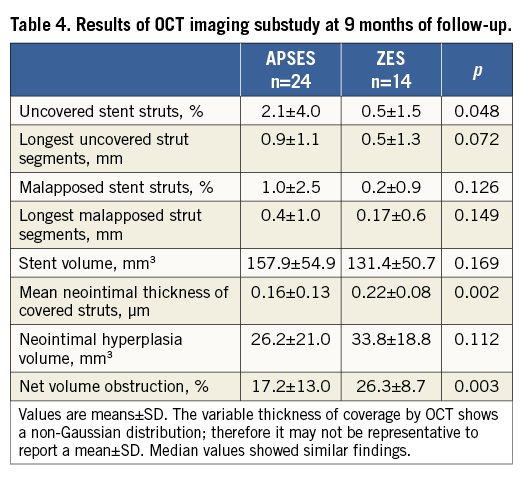
The results of OCT studies, performed at procedure and nine-month follow-up in 24 APSES and 14 ZES patients, are shown in Table 4. The proportion of uncovered stent struts was marginally higher in the APSES (2.1±4.0%) group compared with the ZES (0.5±1.5%) group (p=0.048). The mean neointimal thickness of covered struts was 0.16±0.13 µm in the APSES versus 0.22±0.08 µm in the ZES group (p=0.002). Percent net volume obstruction (17.2±13.0% versus 26.3±8.7%) was also significantly lower in the APSES than in the ZES group (p=0.003). No other significant difference in OCT measurements was observed between the two groups (Table 4).
Adverse clinical events
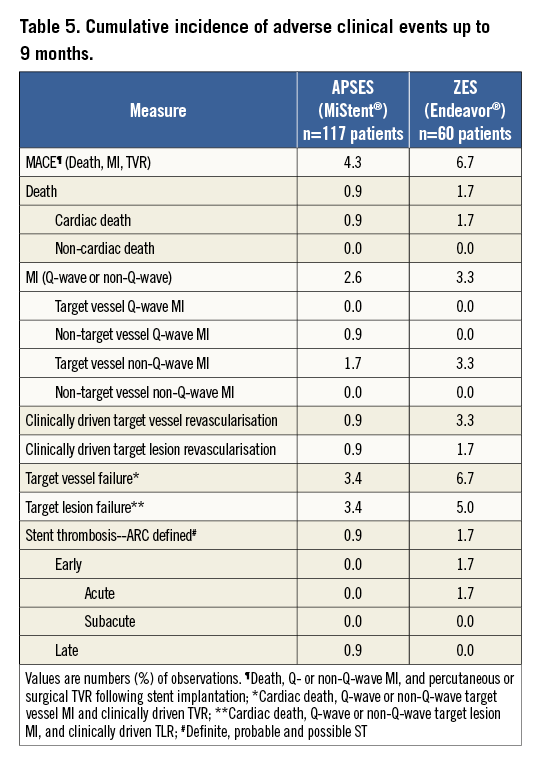
The incidence of in-hospital and long-term adverse clinical events was similar in both groups (Table 5). The overall MACE rate at nine months was 4.3% (5/117) in the APSES and 6.7% (4/60) in the ZES arm; a single cardiac death was observed in each study group and two patients in each group sustained a non-Q-wave MI. The difference in MACE between the two arms was –2.4% with a two-tailed 95% CI from –12.0% to 4.3%. A single definite ST occurred in the ZES group and a single possible ST in the APSES group. These results suggest that the safety profiles of the two study devices are comparable.
Compliance with dual antiplatelet therapy
At six months, 110/119 patients (92.4%) and 47/59 (79.7%) in the APSES and ZES groups, respectively, remained on DAPT. At nine months, the proportions of patients treated with DAPT (83.2% in the APSES versus 79.7% in the ZES groups) were similar.
Discussion
APSES incorporates a proprietary surface modification technology to provide a unique drug delivery system. Compared with previous processes, the drug component is never dissolved during the coating process, thus retaining its crystalline properties. The crystalline sirolimus mitigates an initial burst release of drug and is simultaneously released into the surrounding tissue with the polymer5. The rapid-absorbing polymer formulation is intended to deliver drug into the surrounding tissue, control drug elution and limit the duration of polymer exposure. The drug remains in the tissue well after the polymer is absorbed to moderate any inflammation from the polymer degradation and provides for continued and predictable elution of sirolimus up to nine months. This provides a sustained, long-term inhibition of neointimal proliferation even after the polymer is absorbed, with the potential to avoid the long-term safety concerns and late failures that are associated with durable polymer DES16.
Further to the results of the first-in-human investigation of the APSES8,10,11, this randomised study has confirmed the superior effectiveness of APSES through greater inhibition of neointimal proliferation resulting in a late lumen loss of about half that of ZES. In this trial, while LLL was significantly less for APSES, the TLR rates were similar. In low- to moderate-risk lesions, ZES has also demonstrated comparable TLR rates to TAXUS and CYPHER even though ZES LLL was significantly higher17-19. However, in more complex lesions, high-risk patients or all-comer trials, the TLR rate for ZES is significantly higher than for DES with lower LLL19-22. The DESSOLVE II study design was effective for defining the performance profile of APSES with an LLL value that compares favourably with other clinically effective sirolimus-eluting, zotarolimus-eluting and everolimus-eluting stents which use durable polymers23.
The post-procedure in-stent %DS was the only significant difference at baseline with mean %DS of –0.8% for ZES and 3.4% for APSES (p<0.001). Post-procedure in-lesion %DS was identical in both groups. The mean post-procedure MLD was not different for ZES at 2.9 mm versus 2.8 mm for APSES (p=0.57). There may have been some overexpansion of the ZES stent portending greater LLL. However, mean LLL of 0.58 mm for ZES is very consistent with the 0.58-0.68 mm LLL found in other ZES trials7,17.
While ZES does not strongly inhibit neointimal proliferation, it has been considered a safer DES than TAXUS and CYPHER, providing better vessel “healing” with return of endothelial function24, less acquired incomplete stent apposition and positive remodelling25.
In this study OCT was used in a subgroup of patients to compare in greater detail the vessel response between APSES and ZES. Specific sites were identified to participate in this substudy with enrolled randomised patients consenting to OCT evaluations. For all OCT measurements the two groups were similar except for a significantly lower mean neointimal thickness covering the struts and lower net volume obstruction with APSES. While the percent of uncovered struts was lower for ZES, 2.1% vs 0.5%, both stents had a low percent of uncovered struts. Strut coverage with APSES is at least comparable, if not numerically higher than with other DES evaluated by the same core laboratory26,27.
Study limitations
DESSOLVE II was designed as a single-blind RCT, blinding the patient to treatment assignment. Not blinding the operator could have influenced the operator’s decision about the need for revascularisation. Adverse events were collected at each follow-up visit. At the nine-month visit an ECG was also performed in addition to an assessment of clinical status prior to the angiographic evaluation. To minimise potential bias, an independent CEC was used to adjudicate clinically driven revascularisations.
The results of OCT analysis were reported on per-patient summaries rather than per-strut. While this approach to OCT data analysis may have limitations, this method is commonly used in reporting OCT results and allows comparability with previously reported studies26,27. The main limitation is that lesions with uncovered areas of exactly the same size will weight differently in the analysis depending on the total stented length. Other per-patient approaches, such as the assessment of clusters of malapposed and uncovered struts, can potentially help elucidate better the entire spectrum of stent-vessel interactions12,28,29.
While not evaluated in the DESSOLVE II trial, the rapid absorption of the polymer coating may allow for a reduction in the duration of dual antiplatelet therapy. Whether shortening the device-mandated indication period for DAPT would provide equivalent imaging and clinical results remains to be investigated in larger trials.
Conclusion
The DESSOLVE II randomised clinical trial confirmed the safety and improved efficacy of the APSES compared to a durable polymer-based DES. The primary efficacy endpoint of mean LLL at nine-month angiographic follow-up was superior for APSES compared to ZES, and accompanied by low major adverse cardiac event rates and stent thrombosis in both groups. Long-term clinical outcomes will be followed to five years and an additional larger trial powered for clinical endpoints will be conducted to build on the DESSOLVE II data.
| Impact on daily practice Based on its unique design features, the MiStent absorbable polymer sirolimus-eluting stent shows high antiproliferative properties along with timely vessel healing, both contributing to its demonstrated favourable short-term efficacy/safety profile. Potential for superior long-term biocompatibility of DES with absorbable polymer, hence reduced late failure rates, should be evaluated by long-term follow-up (from 3 to 5 years). In the meantime, further studies should test the clinical performance of the MiStent in various real-life patient and lesion subsets. Opportunities for shortening the duration of mandated DAPT treatment equally deserve further investigation. |
Acknowledgements
Rodolphe Ruffy, MD, contributed to the preparation of this manuscript.
Funding
This trial is sponsored by Micell Technologies, Durham, NC, USA.
Conflict of interest statement
G. Attizani receives consultant fees from St. Jude Medical. D. Donohoe is a consultant and C. Knape is an employee of Micell Technologies. W. Wijns reports institutional grants from several device companies, including Micell Technologies and Medtronic. The other authors have no conflicts of interest to declare.
Online Appendix A. The following investigators and institutions participated in DESSOLVE II
Co-principal Investigators:
William Wijns, Cardiovascular Center Aalst, Aalst, Belgium; John Ormiston, Mercy Angiography, Auckland, New Zealand.
Co-Investigators:
Mathias Vrolix, ZOL (Ziekenhuis Oost-Limburg), Genk, Belgium; Stefan Verheye, Antwerp Cardiovascular Center, ZNA Middelheim, Antwerp, Belgium; Danny Schoors, Brussels University Hospital - UZ Brussel, Brussels, Belgium; Ton Slagboom, OLV, Amsterdam, The Netherlands; Marcel Gosselink, Isala Klinieken, Zwolle, The Netherlands; Edouard Benit, Heart Center Hasselt, Jessaziekenhuis, Hasselt, Belgium; Walter Desmet, KUL Cardiology Gasthuisberg, Leuven, Belgium; Saqib Chowdhary, University Hospital of South Manchester, Manchester, United Kingdom; Maarten Jan Suttorp, St. Antonius Ziekenhuis, Nieuwegein, The Netherlands; Leszek Zagozdzon, Orebro University Hospital, Orebro, Sweden; Marie-Claude Morice, Institut Jacques Cartier – Institut Cardiologique Paris Sud, Massy, France; Carlo Di Mario, Royal Brompton Hospital, Imperial College London, London, United Kingdom; Jean Fajadet, Clinique Pasteur, Toulouse, France; Dougal McClean, Christchurch Hospital, Christchurch, New Zealand; Pieter Stella, University Medical Center (UMC) Utrecht, Utrecht, The Netherlands; Phillipe Garot, Claude Galien Hospital, Quincy, France; Alisdair Ryding, Norfolk and Norwich University Hospital, Norwich, United Kingdom; Iain Simpson, Southampton University Hospital, Southampton, United Kingdom; Jim Stewart, Mercy Angiography Unit, Auckland, New Zealand; Wilbert Aarnoudse, Tweesteden Ziekenhuis, Tilburg, The Netherlands; Per Albertsson, Sahlgrenska University Hospital, Goteborg, Sweden; Cameron Densem, Papworth Hospital, Cambridge, United Kingdom; Scott Harding, Wellington Hospital, Wellington, New Zealand; David Hildick-Smith, Royal Sussex County Hospital, Brighton, United Kingdom; Jim Stewart, Auckland City Hospital, Auckland, New Zealand; Mark Webster, Auckland City Hospital, Auckland, New Zealand.
Core laboratories:
Angiography core laboratory: Alexandra J. Lansky, MD, Yale University School of Medicine, New Haven, CT, USA. Optical Coherence Tomography core laboratory: Marco Costa, MD, PhD, and Hiram Bezerra, MD, PhD, Case Western Medical Center, Cleveland, OH, USA.
Online Appendix B. Main study exclusion criteria
– Myocardial infarction <72 hrs before the index procedure.
– Q-wave or non-Q-wave myocardial infarction ≥72 hrs after DES implant and elevated concentrations of creatine kinase and MB fraction.
– Left ventricular ejection fraction <30% within the past six months.
– Contraindication to dual antiplatelet therapy.
– Direct stenting of the target lesion.
– Target vessel has been treated percutaneously within 10 mm proximal or distal (by visual estimate) of the target lesion in the year preceding the index procedure.
– Planned or completed treatment of the target vessel with an unapproved device, directional or rotational coronary atherectomy, laser, cutting balloon, or transluminal extraction catheter before study stent implantation. History of coronary brachytherapy.
– Coronary angioplasty (with or without stenting) or coronary artery bypass graft surgery planned within nine months or any other intervention planned within 30 days after the index procedure.
– History of percutaneous treatment of a non-target vessel <14 days before study enrolment.
– Cerebrovascular accident or transient ischaemic attack within the past six months.
– Gastrointestinal haemorrhage within past three months.
– Oral or intravenous immunosuppressive therapy or history of life-limiting immunodeficiency or autoimmune disease.
– Serum creatinine concentration >2.0 mg/dl.
– <100,000 platelets/mm³ or <3,000 white blood cells/mm³.
– Suspected or confirmed liver disease.
– Hypersensitivity to sirolimus or structurally related compounds, cobalt-chromium, aspirin, heparin, bivalirudin, clopidogrel, ticlopidine or prasugrel.
– Women of childbearing potential who do not have a negative pregnancy test at baseline or are not on birth control.
– Life expectancy <12 months.
