Abstract
Aims: Our aim was to compare the safety and efficacy of a novel, ultrathin strut, biodegradable polymer sirolimus-eluting stent (BP-SES) with a thin strut, durable polymer everolimus-eluting stent (DP-EES) in a pre-specified subgroup of patients with acute ST-segment elevation myocardial infarction (STEMI) enrolled in the BIOSCIENCE trial.
Methods and results: The BIOSCIENCE trial is an investigator-initiated, single-blind, multicentre, randomised non-inferiority trial (NCT01443104). Randomisation was stratified according to the presence or absence of STEMI. The primary endpoint, target lesion failure (TLF), is a composite of cardiac death, target vessel myocardial infarction, and clinically indicated target lesion revascularisation within 12 months. Between February 2012 and May 2013, 407 STEMI patients were randomly assigned to treatment with BP-SES or DP-EES. At one year, TLF occurred in seven (3.4%) patients treated with BP-SES and 17 (8.8%) patients treated with DP-EES (RR 0.38, 95% CI: 0.16-0.91, p=0.024). Rates of cardiac death were 1.5% in the BP-SES group and 4.7% in the DP-EES group (RR 0.31, 95% CI: 0.08-1.14, p=0.062); rates of target vessel myocardial infarction were 0.5% and 2.6% (RR 0.18, 95% CI: 0.02-1.57, p=0.082), respectively, and rates of clinically indicated target lesion revascularisation were 1.5% in the BP-SES group versus 2.1% in the DP-EES group (RR 0.69, 95% CI: 0.16-3.10, p=0.631). There was no difference in the risk of definite stent thrombosis.
Conclusions: In this pre-specified subgroup analysis, BP-SES was associated with a lower rate of target lesion failure at one year compared to DP-EES in STEMI patients. These findings require confirmation in a dedicated STEMI trial.
Introduction
Acute ST-segment elevation myocardial infarction (STEMI) confers an increased risk of adverse outcome compared to stable coronary artery disease which extends beyond the periprocedural phase of primary percutaneous coronary intervention (PCI). Plaque characteristics of culprit lesions, thrombus burden and persistent inflammation are hallmarks of STEMI patients which increase the risk of delayed arterial healing and vessel remodelling, as reflected by higher rates of incomplete stent strut coverage1,2 and malapposition3.
Biodegradable polymer-based metallic drug-eluting stents (BP-DES) have been observed to reduce the rate of major adverse cardiac events (MACE) compared to bare metal stents (BMS) among patients with STEMI undergoing primary PCI at one year4. BP-DES have also been associated with a favourable healing response in intravascular imaging studies5, and demonstrated a lower incidence of MACE throughout long-term follow-up compared to early-generation durable polymer drug-eluting stents among patients with STEMI6,7. A randomised comparison of durable polymer everolimus-eluting stents (DP-EES) versus BMS failed to show superiority of DP-EES with regard to the patient-oriented primary composite endpoint of all-cause death, recurrent myocardial infarction, and repeat revascularisation at one year. However, rates of target lesion revascularisation and definite and probable stent thrombosis were significantly lower in patients treated with DP-EES compared with BMS8.
New-generation DES with cobalt-chromium platforms, reduced stent strut thickness and biocompatible polymers have been shown to be safe and effective in unselected patient populations and represent the current standard of care in patients undergoing PCI9. The combination of biodegradable polymers with thin strut cobalt-chromium platforms represents the next iteration of technological progress. In the randomised controlled BIOSCIENCE (biodegradable polymer sirolimus-eluting stent versus durable polymer everolimus-eluting stent for percutaneous coronary revascularisation) trial, an ultrathin strut cobalt-chromium stent releasing sirolimus from a biodegradable poly-L-lactic acid (PLLA) polymer (BP-SES) was non-inferior compared to a thin strut DP-EES in a patient population reflecting routine clinical practice10. We present the results of a pre-specified subgroup analysis of patients with STEMI enrolled into the BIOSCIENCE trial.
Methods
STUDY DESIGN AND PATIENTS
The BIOSCIENCE trial is an investigator-initiated, single-blind, multicentre, randomised non-inferiority trial comparing an ultrathin strut (60 μm for stent diameters up to 3.0 mm, 80 μm for stent diameters >3.0 mm) cobalt-chromium L605 stent platform covered with an amorphous silicon-carbide layer and a biodegradable PLLA polymer releasing sirolimus (Orsiro; Biotronik AG, Bülach, Switzerland) with a thin strut cobalt-chromium, durable polymer everolimus-eluting stent (XIENCE PRIME/Xpedition®; Abbott Vascular, Santa Clara, CA, USA) in a patient population with minimal exclusion criteria in nine centres in Switzerland. In summary, all patients aged 18 years or older with at least one coronary lesion with >50% diameter de novo stenosis or restenosis in a native coronary artery or a bypass graft suitable for stent implantation were eligible for inclusion. There were no restrictions in terms of number of vessels or lesions treated. Inclusion and exclusion criteria as well as detailed characteristics of the study devices have been reported previously11. The study was approved by the institutional ethics committees of all participating sites and complied with the Declaration of Helsinki. All study participants provided written informed consent. The trial is registered with ClinicalTrials.gov (NCT01443104). The trial was supported by an unrestricted grant from Biotronik, Bülach, Switzerland. The funding source had no role in the study design, data collection, data monitoring, data analysis, or data interpretation.
RANDOMISATION
Patients were randomly allocated in a 1:1 ratio to BP-SES or DP-EES immediately after diagnostic angiography and prior to PCI. Randomisation was computer generated and stratified according to the presence or absence of STEMI, per site. Sequentially numbered, opaque, sealed envelopes were used as a back-up in case of malfunction of the web-based randomisation system.
PROCEDURES
PCI was performed according to current guidelines. Thrombus aspiration, predilation, and post-dilatation were performed according to the discretion of the operator. Staged repeat revascularisation procedures with the assigned study stent were scheduled within three months of the index procedure. Patients were treated with unfractionated heparin with a dose of 5,000 IU or 70-100 IU/kg body weight during the procedure. The administration of bivalirudin instead of unfractionated heparin was allowed. Administration of glycoprotein IIb/IIIa inhibitors was left to the discretion of the operator. Antiplatelet therapy was initiated upstream or at the time of primary PCI. Acetylsalicylic acid (≥250 mg) was combined with clopidogrel (loading dose 600 mg, maintenance dose 75 mg QD), prasugrel (loading dose 60 mg, maintenance dose 10 mg QD), or ticagrelor (loading dose 180 mg, maintenance dose 90 mg BID). After a recommended dual antiplatelet therapy of 12 months duration, monotherapy with acetylsalicylic acid was continued indefinitely.
DEFINITIONS AND DATA MANAGEMENT
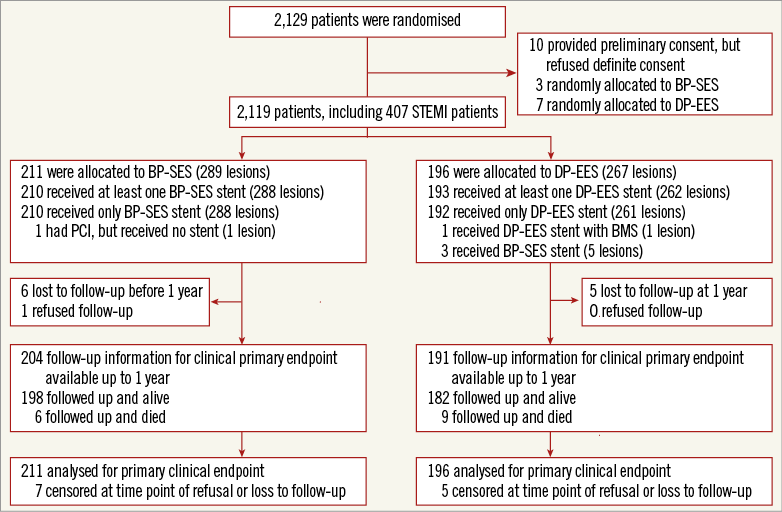
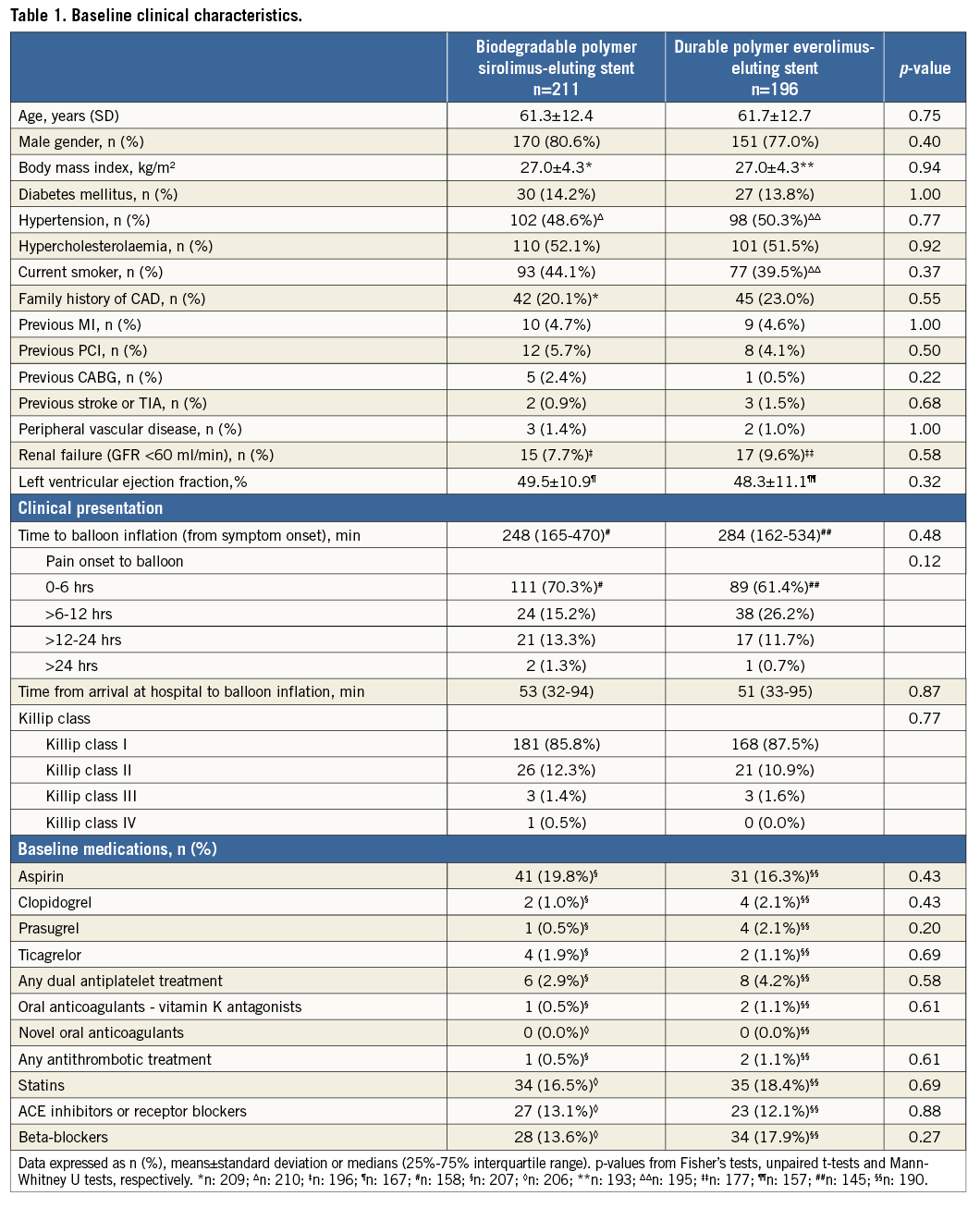
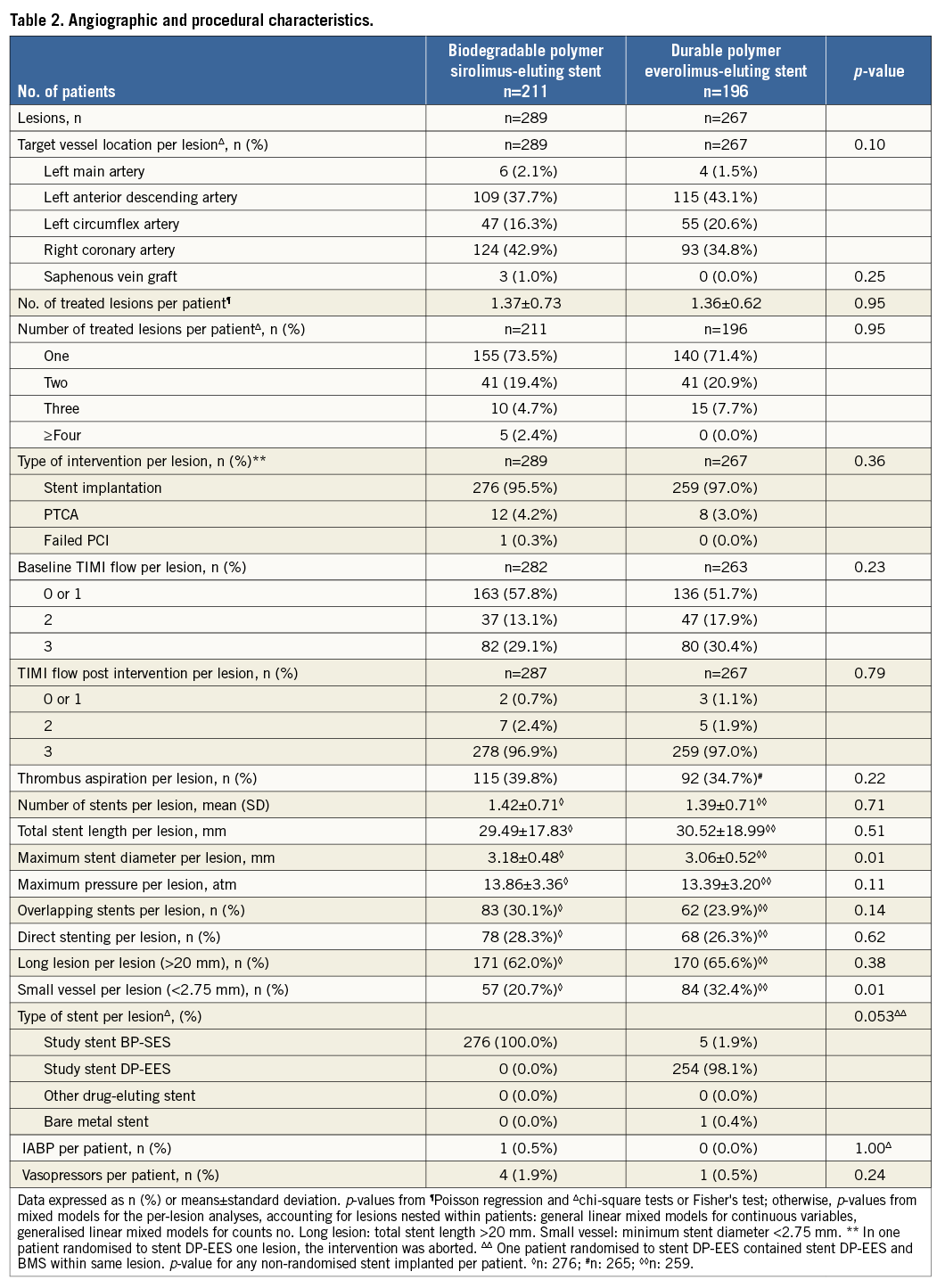
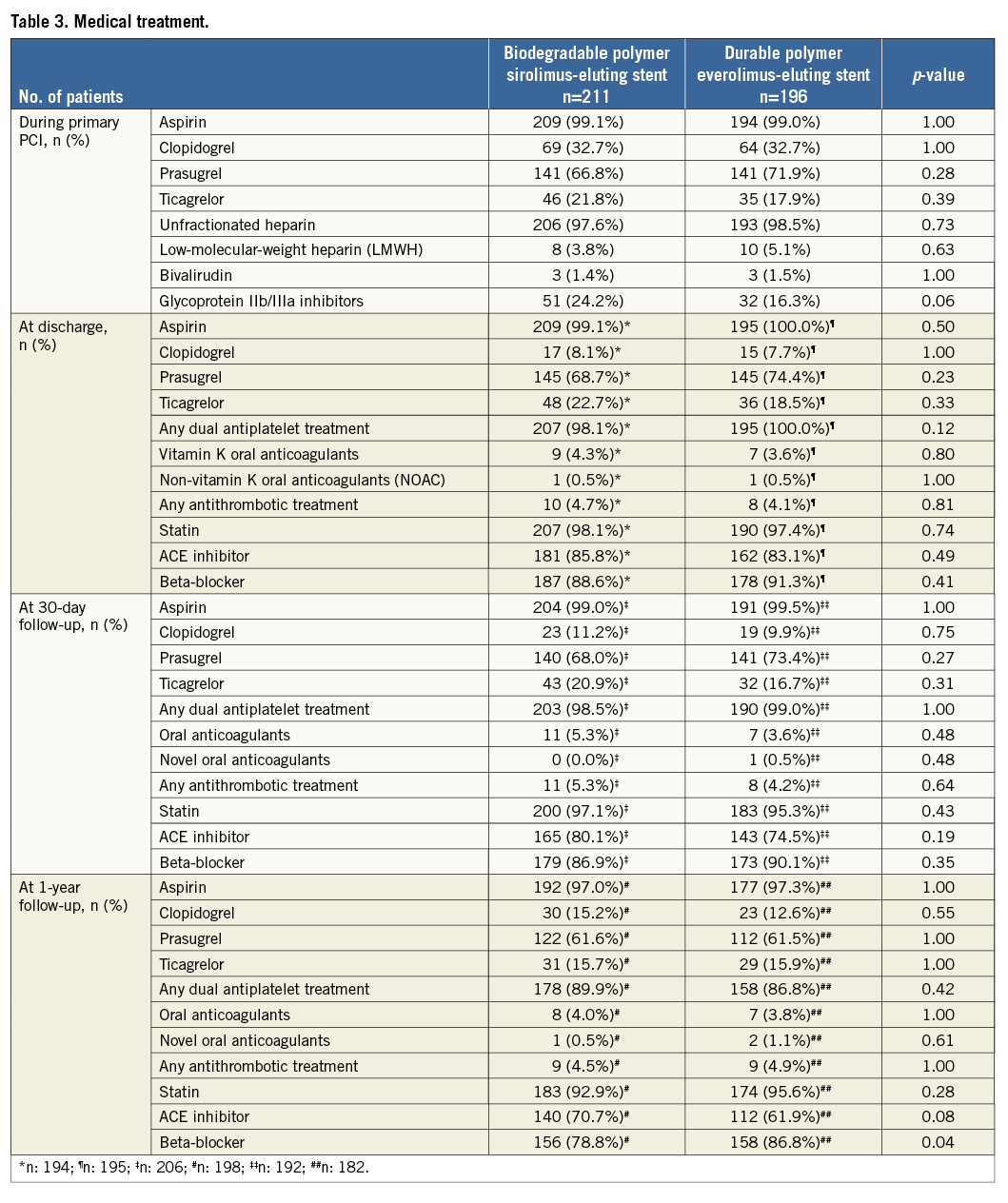
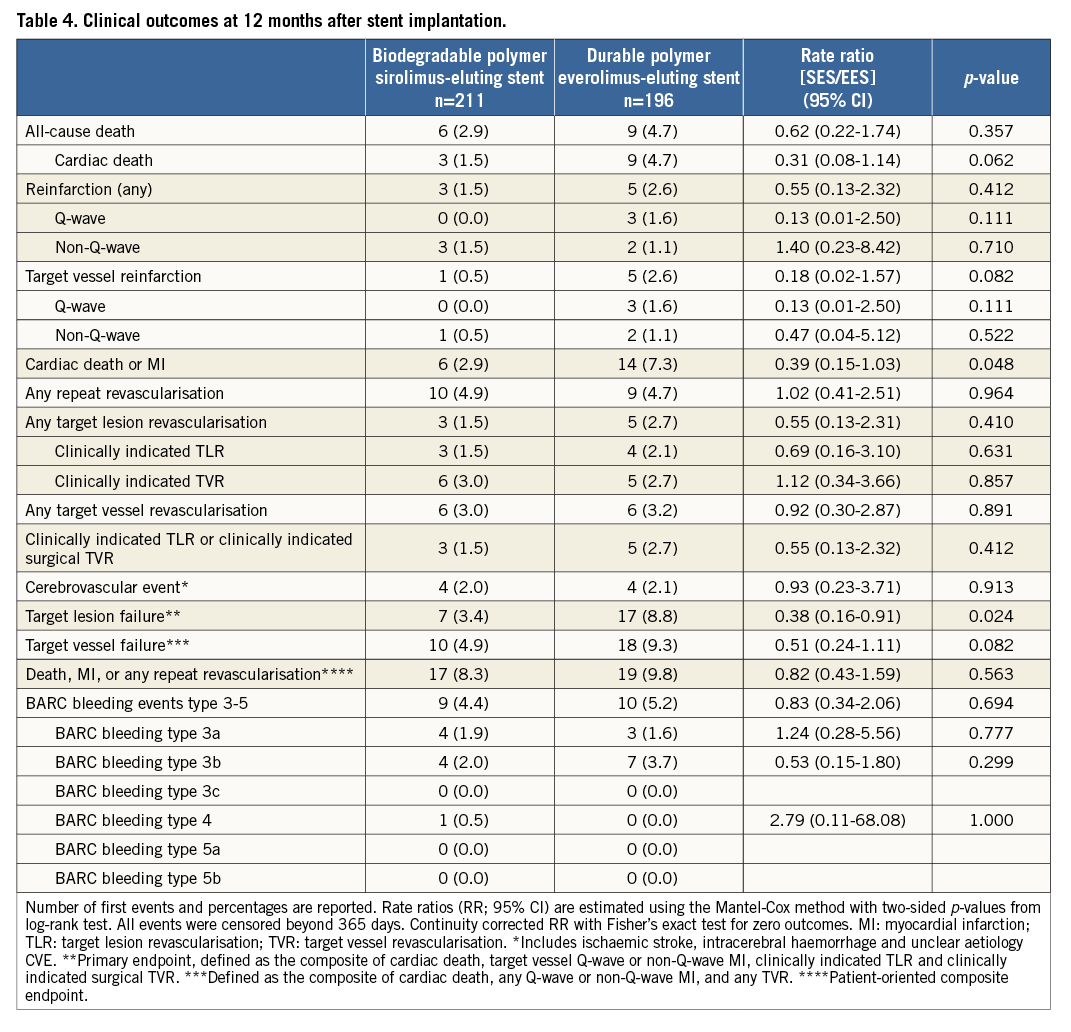
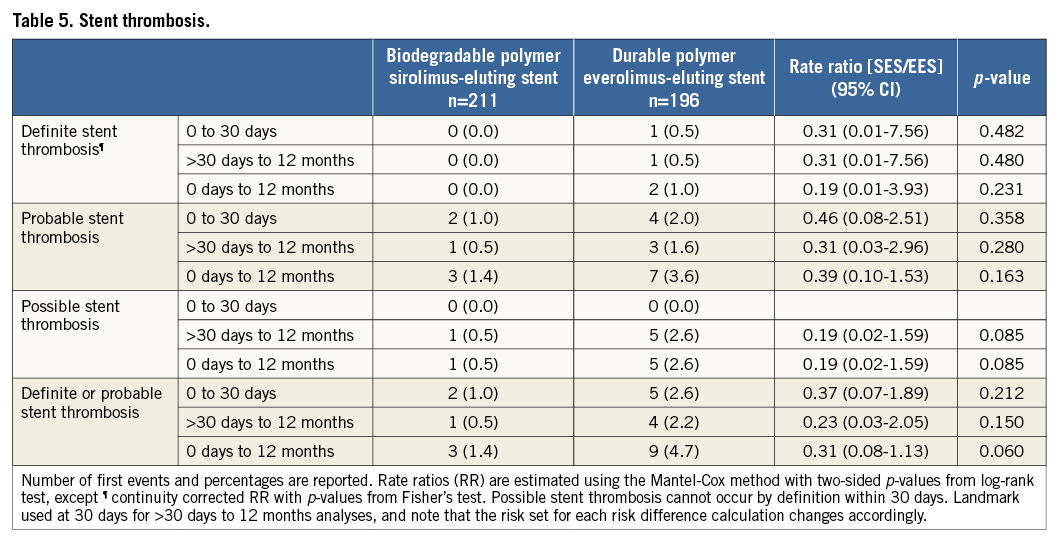
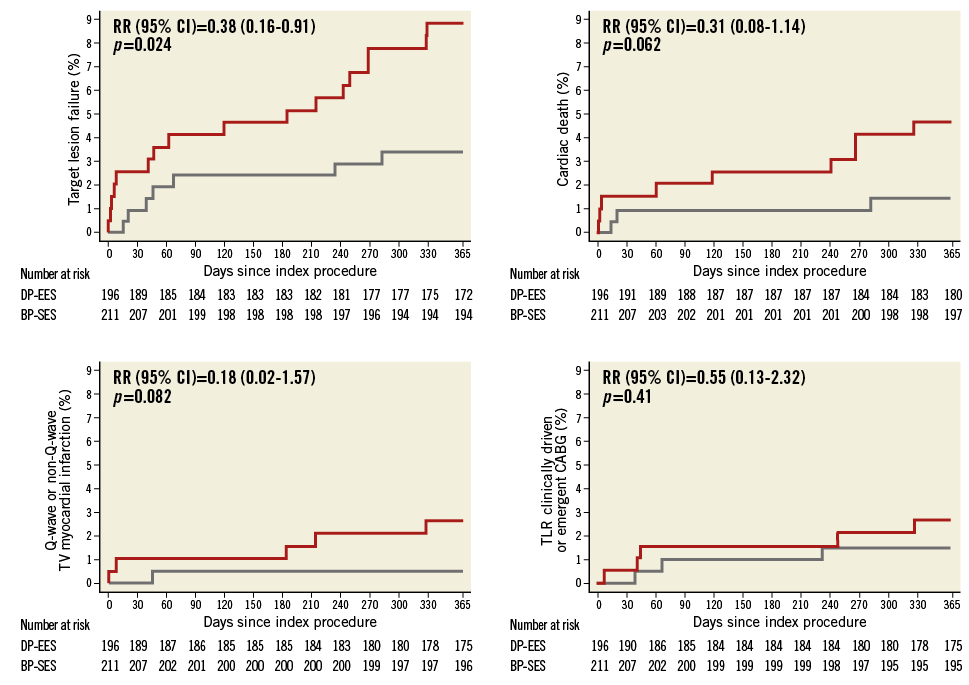
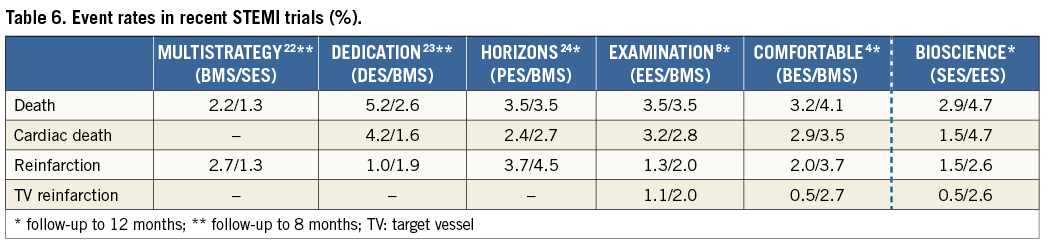
Definitions of primary and secondary endpoints have been outlined previously11. Target lesion failure was the pre-specified primary endpoint and was a composite of cardiac death, target vessel myocardial infarction, and clinically indicated target lesion revascularisation (TLR) within 12 months. Cardiac death was defined as any death due to an immediate cardiac cause, death related to the procedure, unwitnessed death, and death of unknown cause. Spontaneous myocardial infarction was recorded in case of a typical rise and fall of creatinine kinase MB fraction or troponin in the presence of at least one of the following: ischaemic symptoms, new pathologic Q-waves, ischaemic electrocardiographic changes or pathological evidence of acute myocardial infarction. Periprocedural myocardial reinfarction in the setting of evolving myocardial infarction was defined as recurrent chest pain lasting >20 minutes (or new ECG changes consistent with myocardial infarction) in combination with a >50% increase of peak CK (or CK-MB in the absence of CK) level above the previous level measured within 24 hours after the event. In case elevated CK (or CK-MB) levels from the index infarction were falling or had returned to normal, a reinfarction was diagnosed in case of new elevation of CK >2x the upper limit of normal (ULN) if the CK level had returned to All data were entered into a web database held at the Clinical Trials Unit and the Department of Cardiology in Bern, Switzerland. Regular follow-up was performed at 30 days and one year. Electrocardiograms were systematically recorded at baseline, after the procedure, at 12-month follow-up and in case of recurrent signs or symptoms of ischaemia. Data monitoring and event adjudication have been described previously. STATISTICAL ANALYSIS The BIOSCIENCE trial was powered for non-inferiority on the primary clinical endpoint, target lesion failure at 12 months, in the overall population but not in the pre-specified subgroup of STEMI patients. Based on event rates reported from COMPARE12, RESOLUTE All-Comers9, and the LESSON registry13, a TLF rate of 8% at 12 months was assumed in both treatment arms. Using a margin of 3.5% for non-inferiority of BP-SES vis-à-vis DP-EES, enrolment of 2,060 patients was calculated to provide at least 80% power to detect non-inferiority at a one-sided type I error of 0.05. The STEMI population was a pre-specified subgroup with a significant interaction effect (STEMI yes or no vs. randomised stent) reported for the primary outcome10, which warranted a more in-depth assessment of clinical outcomes reported here. Clinical endpoints were analysed according to the intention-to-treat principle. We used the Mantel-Cox method to calculate rate ratios (RR), two-sided 95% confidence intervals (CI) and corresponding two-sided p-values for superiority from the log-rank test. We used time to first event for each type of outcome throughout, and report Kaplan-Meier estimates of event rates. All analyses of endpoints were performed according to the intention-to-treat principle. Analyses were undertaken by a statistician of the Clinical Trials Unit Bern in Stata version 13 (StataCorp LP, College Station, TX, USA). All p-values and CIs are reported two-sided. P-values for characteristics recorded at the patient level are from unpaired t-tests, chi-square tests, or Fisher’s exact tests, except when specified. P-values for characteristics that were recorded at the lesion level are from general or generalised linear mixed models to account for the non-independence of lesions within the same patient. Results Among 2,119 patients randomly assigned to treatment with BP-SES or DP-EES between February 2012 and May 2013, 407 patients (19%) presented with STEMI. Two hundred and eleven STEMI patients with 289 lesions were allocated to treatment with BP-SES, and 196 STEMI patients with 267 lesions were allocated to treatment with DP-EES (Figure 1). Figure 1. Patient flow according to the CONSORT statement. BP-SES: biodegradable polymer sirolimus-eluting stent; DP-EES: durable polymer everolimus-eluting stent Baseline clinical characteristics were comparable between the two treatment arms (Table 1). The time interval from pain onset to presentation was ≤6 hours in 111 (70%) patients with BP-SES and 89 (61%) patients with DP-EES. The median door-to-balloon time amounted to 53 (interquartile range [IQR] 32-93) minutes in the BP-SES group and 51 (IQR 33-95) minutes in the DP-EES group, respectively. More than 98% of patients were in Killip class I or II in both treatment groups at the time of primary PCI. Angiographic and procedural features were similar between the two treatment arms with the exception of a higher number of small vessels per lesion among patients treated with DP-EES as compared with BP-SES (84 [32%] versus 57 [21%], p=0.01) and a smaller maximal stent diameter (3.1±0.5 mm versus 3.2±0.5 mm, p=0.01), respectively (Table 2). Medical treatment during the procedure and throughout follow-up is summarised in Table 3 without significant differences. There were no differences in periprocedural antiplatelet and antithrombotic therapy between groups. The majority of patients were loaded with a novel P2Y12 inhibitor primarily, or in addition to clopidogrel. Glycoprotein IIb/IIIa inhibitors were used in 24.2% of patients treated with BP-SES and 16.3% of patients treated with DP-EES (p=0.06). Periprocedural antithrombotic treatment consisted of unfractionated heparin in the overwhelming majority of patients. Clinical outcome is summarised in Table 4 and illustrated in Figure 2. Among STEMI patients, the primary endpoint TLF occurred in seven (3.4%) patients treated with BP-SES and 17 (8.8%) patients treated with DP-EES at one year (RR 0.38, 95% CI: 0.16-0.91, p=0.024). The rates of the individual components of the composite endpoint TLF are summarised in Figure 2. There was a non-significant trend towards lower rates of cardiac death (BP-SES three [1.5%] versus DP-EES nine [4.7%], RR 0.31, 95% CI: 0.08-1.14, p=0.062) and target vessel myocardial infarction (BP-SES one [0.5%] versus DP-EES five [2.6%], RR 0.18, 95% CI: 0.02-1.57, p=0.082) in patients treated with BP-SES. Rates of clinically indicated TLR were 1.5% in the BP-SES group versus 2.1% in the DP-EES group (RR 0.69, 95% CI: 0.16-3.10, p=0.631). Rates of definite, probable, and possible stent thrombosis (ST) at different time points are summarised in Table 5. Whereas there were no differences in rates of definite ST, rates of definite or probable ST were numerically more frequent in the DP-EES treatment arm at one year. Figure 2. Time-to-event curves for the composite endpoint target lesion failure and individual components of the primary endpoint up to 12 months of follow-up. A) Target lesion failure. B) Cardiac death. C) Target vessel (TV) myocardial infarction. D) Clinically indicated target lesion revascularisation (TLR) or emergent coronary artery bypass surgery (CABG). Blue lines indicate BP-SES, red lines indicate DP-EES. Discussion In the pre-specified subgroup analysis of the randomised BIOSCIENCE trial, BP-SES was associated with a lower rate of the primary endpoint TLF at one year compared to DP-EES. Differences in the primary endpoint were driven by non-significant, numerical differences in cardiac death and target vessel myocardial infarction, whereas there was no difference in repeat revascularisation. The antirestenotic efficacy of early-generation durable polymer sirolimus-eluting stents (DP-SES) in the setting of STEMI came at the expense of an excess in late thrombotic events14. In the LEADERS (Limus Eluted from A Durable versus ERodable Stent coating) trial15, which compared biodegradable polymer biolimus-eluting stents (BP-BES) with early-generation DP-SES, a lower rate of the primary endpoint MACE among BP-BES treated patients was documented in the subgroup of STEMI at nine months, a finding which was maintained during long-term follow-up throughout five years16. The present study may extend the potential benefit of biodegradable polymer-based DES among STEMI patients observed with early-generation thick-strut stainless steel DES to newer-generation thin-strut cobalt-chromium DES. Noteworthy, cobalt-chromium DP-EES currently represent the benchmark for safety and efficacy in STEMI patients undergoing primary PCI, as suggested by recent network meta-analyses17,18. An individual patient-data, pooled analysis of 497 patients with STEMI from three trials randomly comparing thick-strut stainless steel BP-DES with early-generation, thick-strut DP-SES observed a benefit of BP-DES with a difference emerging within one year after primary PCI (BP-DES 27 [9.4%] versus early-generation DP-SES 32 [15.8%], HR 0.58, 95% CI: 0.35-0.93, p=0.03), whereas there was no difference in the period between one and four years (BP-DES 13 [5.2%] versus early-generation DP-SES 14 [8.6%], HR 0.62, 95% CI: 0.29-1.31, p=0.21). In contrast to the present analysis, the effect within the first year was driven by a lower rate of repeat revascularisation (TLR: BP-DES 12 [4.4%] versus early-generation DP-SES 19 [9.7%], HR 0.43, 95% CI: 0.21-0.89, p=0.02), while no difference was observed in rates of cardiac death or myocardial infarction6. A trend towards a lower rate of definite stent thrombosis in the first year after stent implantation documented in the pooled analysis (BP-DES eight [2.9%] versus early-generation DP-SES 13 [6.5%], HR 0.43, 95% CI: 0.18-1.03, p=0.06) was also seen in the present subgroup analysis6. Five-year outcomes of the LEADERS trial showed significantly improved safety and efficacy outcomes in patients with STEMI treated with BP-BES as compared to early-generation DP-SES, driven by a lower rate of cardiac death (BP-BES four [3%] versus DP-SES 16 [11.4%], p=0.007) and revascularisation (BP-BES 14 [17.8%] versus 37 [26.4%], p=0.049)7. It remains to be determined whether these findings are a class effect of biodegradable polymer DES in general, or whether the properties are inherent to individual stents specifically. Biodegradable polymers may have favourable results on arterial healing after DES implantation. In the optical coherence tomography substudy of the LEADERS trial5, a favourable healing response was observed with BP-BES compared with early-generation DP-SES at nine months. The effect of an enhanced healing response may be augmented in the inflammatory milieu of STEMI. Evidence of adverse arterial remodelling among patients with STEMI has been shown by a greater degree of incomplete DES apposition and delayed tissue coverage compared to patients with stable or unstable angina in optical coherence tomography studies1,19. However, while sirolimus is released over a period of 12 to 14 weeks, the PLLA polymer matrix degrades over a period of 12 to 24 months, and may not fully account for the observed difference. In addition to the biodegradable polymer, the reduced strut thickness of the novel stent platform as well as the passive stent coating may play a role when implanted into lesions of patients with STEMI, by reducing acute arterial injury and the risk for peripheral embolisation as well as affording a more rapid re-endothelialisation. A reduction of strut thickness in the case of BMS has been associated with a lower risk of restenosis20 and attenuated thrombogenicity21. Limitations The present analysis has several limitations. First, the BIOSCIENCE trial was not powered to assess differences in clinical outcome among patients with STEMI and therefore findings may be due to chance alone. However, randomisation was pre-specified for the presence or absence of STEMI and there were no differences in evidence-based medical therapy. Furthermore, event rates were consistent with event rates of recent STEMI trials (Table 6). Second, patients in the DP-EES treatment arm had a higher proportion of small vessel disease and a smaller maximum stent diameter that may have affected clinical outcome. Moreover, there was a trend towards a more frequent administration of glycoprotein IIb/IIIa inhibitors in the group of patients treated with BP-SES. Third, the STEMI patients included in the BIOSCIENCE trial were rather low risk based on Killip class; generalisability to a higher-risk population therefore requires confirmation in registries including higher-risk patients. Finally, the duration of follow-up was limited to 12 months, which may not suffice to assess the efficacy of BP-SES beyond complete degradation of the biodegradable polymer. Conclusion In conclusion, BP-SES may be associated with improved clinical outcomes compared with DP-EES among STEMI patients undergoing primary PCI. The findings are hypothesis-generating and have to be reproduced in a dedicated, randomised trial in order to substantiate the potential benefit of BP-SES in patients with STEMI. Impact on daily practice The use of a thin-strut biodegradable polymer sirolimus-eluting stent (Orsiro BP-SES) in patients with acute ST-segment elevation myocardial infarction (STEMI) is safe and effective. In this pre-specified analysis of the BIOSCIENCE trial, the BP-SES had a similar performance compared with the durable polymer everolimus-eluting stents, with a significant reduction in the risk of target lesion failure. This finding needs confirmation at long-term follow-up and warrants further clinical evaluation in an appropriate randomised trial. However, the Orsiro BP-SES is currently a reasonable alternative when implanted in the STEMI setting. Appendix. Contributors T. Pilgrim, R. Piccolo, D. Heg, P. Jamshidi and S. Windecker conceived the study. T. Pilgrim, D. Heg, P. Jamshidi, and S. Windecker had responsibility for the design of the study. T. Pilgrim, R. Piccolo, D. Heg, M. Roffi, D. Tüller, A. Vuilliomenet, O. Muller, S. Cook, D. Weilenmann, C. Kaiser, A. Khattab, M. Taniwaki, F. Rigamonti, S. Blöchlinger, P. Wenaweser, P. Jüni, S. Windecker were responsible for the acquisition of data. D. Heg did the analysis and interpreted the results in collaboration with T. Pilgrim, R. Piccolo, P. Jüni, S. Windecker and all other authors. T. Pilgrim, R. Piccolo, D. Heg and S. Windecker wrote the first draft of the report. All authors critically revised the report for important intellectual content and approved the final version. Funding Funding for the current analysis was provided by the Clinical Trials Unit, University of Bern, and Biotronik, Bülach, Switzerland. Conflict of interest statement T. Pilgrim has received travel expenses and payment for lectures from Biotronik and Medtronic. R. Piccolo received a research grant from Veronesi Foundation. M. Roffi has received grants from Boston Scientific, Abbott Vascular, Medtronic, and Biosensors, and payment for lectures from Lilly-Daiichi Sankyo. D. Tüller has received travel expenses from Biotronik, Biosensors, Terumo, and Medtronic. S. Cook has received grants and personal fees from Boston Scientific, grants from Medtronic and Cordis, and personal fees from St. Jude Medical. C. Kaiser has received grants from B. Braun, Biotronik, Abbott Vascular, Terumo, Daiichi Sankyo, Eli Lilly, personal fees from Eli Lilly, AstraZeneca, Abbott Vascular, GE Healthcare, Eli Lilly, Daiichi Sankyo. P. Wenaweser has received personal fees from Biotronik and Cordis, grants and personal fees from Medtronic, Edwards Lifesciences, Symetis, and Boston Scientific, and grants from JenaValve, NVT and St. Jude Medical. P. Jüni is an unpaid steering committee or statistical executive committee member of trials funded by Abbott Vascular, Biosensors, Medtronic and Johnson & Johnson. CTU Bern, which is part of the University of Bern, has a staff policy of not accepting honoraria or consultancy fees. However, CTU Bern is involved in design, conduct, or analysis of clinical studies funded by Abbott Vascular, Ablynx, Amgen, AstraZeneca, Biosensors, Biotronik, Boehringer Ingelheim, Eisai, Eli Lilly, Exelixis, Geron, Gilead Sciences, Nestlé, Novartis, Novo Nordisc, Padma, Roche, Schering-Plough, St. Jude Medical, and Swiss Cardio Technologies. S. Windecker has received research contracts to the institution from Biotronik and St. Jude. The other authors have no conflicts of interest to declare.