
Abstract
Aims: A novel ultrathin-strut biodegradable polymer sirolimus-eluting stent (BP-SES) (Orsiro; Biotronik, Bülach, Switzerland) was shown to be superior to a thin-strut durable polymer everolimus-eluting stent (DP-EES) (XIENCE Xpedition/Alpine; Abbott Vascular, Santa Clara, CA, USA) with respect to the primary endpoint of target lesion failure (TLF) at 12 months in the pre-specified subgroup of patients with ST-segment elevation myocardial infarction (STEMI) included in the BIOSCIENCE trial. We designed a large-scale, randomised, clinical trial to assess the clinical superiority of ultrathin-strut BP-SES over thin-strut DP-EES among patients with STEMI undergoing primary percutaneous coronary intervention (PPCI).
Methods and results: BIOSTEMI (NCT02579031) is a prospective, multicentre, randomised, controlled, superiority trial that will randomly assign 1,250 patients with STEMI undergoing PPCI to treatment with BP-SES or DP-EES. The primary endpoint of TLF, a composite of cardiac death, target vessel reinfarction, and clinically indicated target lesion revascularisation within 12 months, will be analysed with Bayesian models applied to the BIOSTEMI data set (n=1,250) using robust historical priors to incorporate historical information from the BIOSCIENCE STEMI subgroup (n=407).
Conclusions: The BIOSTEMI trial will determine whether ultrathin-strut BP-SES are superior to thin-strut DP-EES with respect to TLF in patients with STEMI undergoing PPCI.
Abbreviations
BMS: bare metal stent
BP-DES: biodegradable polymer drug-eluting stent
BP-SES: biodegradable polymer sirolimus-eluting stent
DES: drug-eluting stent
DP-DES: durable polymer drug-eluting stent
DP-EES: durable polymer everolimus-eluting stent
MACE: major adverse cardiac events
PCI: percutaneous coronary intervention
RR: rate ratio
STEMI: ST-segment elevation myocardial infarction
ST: stent thrombosis
TLF: target lesion failure
TLR: target lesion revascularisation
TVR: target vessel revascularisation
Introduction
Primary percutaneous coronary intervention (PPCI) is the reperfusion strategy of choice for patients with acute ST-segment elevation myocardial infarction (STEMI)1. However, PPCI during STEMI may predispose to stent malapposition due to stent undersizing and later thrombus resolution, thereby increasing the risk of stent thrombosis (ST) and in-stent restenosis2. In addition, vessel healing at the culprit site of STEMI is substantially delayed and may result in increased rates of late ST3. In this setting, early-generation durable polymer drug-eluting stents (DP-DES) were associated with substantial reductions in target vessel revascularisation (TVR) rates compared with bare metal stents (BMS)4,5, but this early benefit was offset by an increased risk of very late ST during subsequent years4,5, related to the persistence of durable polymers that results in chronic local inflammation, impaired endothelialisation and delayed arterial healing3.
Newer-generation drug-eluting stents (DES) with thinner-strut stent platforms, biocompatible permanent polymer coatings and reduced sirolimus-analogue drug loads were associated with improved vascular healing and clinical outcomes compared with early-generation DP-DES6. Among patients with STEMI, the thin-strut durable polymer everolimus-eluting stent (DP-EES) was shown to reduce rates of repeat revascularisation and ST compared with BMS at one-7 and two-year8 follow-up, without increasing the risk of cardiac death or myocardial infarction (MI). Nevertheless, several randomised trials consistently failed to demonstrate clinical superiority of newer-generation biocompatible DP-DES over early-generation thick-strut DP-DES in STEMI patients treated with PPCI9-12.
Newer-generation biodegradable polymer DES (BP-DES) were recently developed to overcome delayed vascular healing attributed to chronic inflammatory responses13 and hypersensitivity reactions14 induced by permanent polymer coatings, that result in persisting late adverse clinical events15. Newer-generation BP-DES were shown to be a safe and effective alternative to DP-DES in numerous randomised trials16-24 and large-scale comprehensive network meta-analyses25-27. However, randomised evidence on the clinical efficacy and safety of BP-DES in STEMI patients undergoing PPCI is scarce. In the Comparison of Biolimus Eluted From an Erodible Stent Coating With Bare Metal Stents in Acute ST-Elevation Myocardial Infarction (COMFORTABLE AMI) trial, thick-strut biodegradable polymer biolimus-eluting stents (BP-BES) were associated with reduced short-28 and long-term29 rates of major adverse cardiac events (MACE) compared with BMS, mainly driven by lower risk of target vessel reinfarction and ischaemia-driven TLR. In a recent subgroup analysis of the randomised Limus Eluted From A Durable Versus ERodable Stent Coating (LEADERS) trial, thick-strut BP-BES were shown to reduce rates of MACE, cardiac death and definite ST throughout five years compared with early-generation thick-strut DP-DES30. These findings were recently confirmed by a pooled individual patient-level data analysis demonstrating significant reductions in rates of MACE and TLR at four years among STEMI patients treated with newer-generation thick-strut BP-BES compared with early-generation thick-strut DP-DES31. However, no randomised evidence is currently available comparing BP-DES with newer-generation DP-DES for PPCI.
The Orsiro DES (Biotronik, Bülach, Switzerland) is a novel biodegradable polymer sirolimus-eluting stent (BP-SES) consisting of an ultrathin-strut cobalt-chromium metallic stent platform and a unique hybrid combination of passive and active coatings32. The thin-layer, amorphous, silicon-carbide passive coating encapsulates the stent surface to reduce ion release from the metal stent platform and therefore minimises interaction with the surrounding tissue. The active coating contains a biodegradable poly-L-lactic acid (PLLA) polymer matrix that degrades completely within approximately 12-15 months and delivers sirolimus at a dose of 1.4 μg/mm2 with elution kinetics optimised to release in vivo over approximately 100 days32. In the Ultrathin-strut Biodegradable Polymer Sirolimus-Eluting Stent Versus Durable Polymer Everolimus Eluting Stent for Percutaneous Coronary Revascularisation (BIOSCIENCE) randomised trial, ultrathin-strut BP-SES were shown to be non-inferior to current best-in-class thin-strut biocompatible DP-EES with respect to the primary composite endpoint of target lesion failure (TLF) at one- and two-year follow-up22,33 in all-comers patients undergoing PCI. In the pre-specified subgroup of patients with STEMI, ultrathin-strut BP-SES were associated with significantly reduced rates of TLF during short- and long-term follow-up34,35 compared with thin-strut DP-EES. Based on these hypothesis-generating findings, we designed a large-scale, randomised trial to assess the clinical superiority of ultrathin-strut BP-SES over thin-strut DP-EES in patients with STEMI undergoing PPCI.
Methods
STUDY DESIGN AND PRIMARY HYPOTHESIS
BIOSTEMI (A Comparison of an Ultrathin Strut Biodegradable Polymer Sirolimus-Eluting Stent With a Durable Polymer Everolimus-Eluting Stent for Patients With Acute ST-Segment Elevation Myocardial Infarction Undergoing Primary Percutaneous Coronary Intervention) is a prospective, multicentre, assessor-blind, randomised, controlled trial comparing the ultrathin-strut Orsiro BP-SES with the state-of-the-art thin-strut biocompatible DP-EES (XIENCE Xpedition®/Alpine™; Abbott Vascular, Santa Clara, CA, USA) in patients with STEMI undergoing PPCI (Figure 1). The study protocol was designed by the steering committee, and all data are managed by the Clinical Trials Unit (CTU) Bern, University of Bern, Switzerland. The study organisation is detailed in Supplementary Table 1. The trial is registered at www.clinicaltrials.gov (Identifier: NCT02579031). The first patient was included in April 2016.
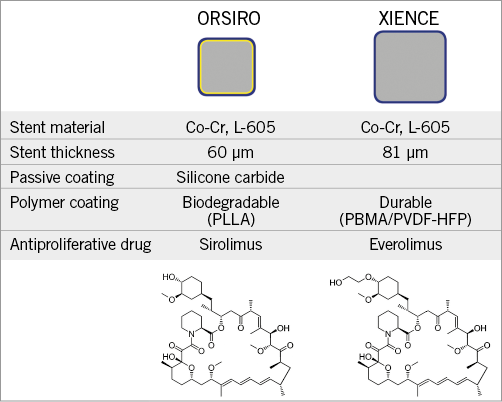
Figure 1. Overview of the principal characteristics of the study stents. Co: cobalt; Cr: chromium; PBMA: poly n-butyl methacrylate; PLLA: poly-L-lactic acid; PVDF-HFP: poly-vinylidene fluoride-co-hexafluoropropylene
The primary objective of the BIOSTEMI trial is to investigate the study hypothesis that ultrathin-strut BP-SES are superior to thin-strut DP-EES in STEMI patients undergoing PPCI. The study will combine data from the previously initiated BIOSCIENCE study22 with prospective evidence from the BIOSTEMI trial using a robust Bayesian approach (Figure 2). The main features of the BIOSCIENCE and BIOSTEMI study designs and inclusion criteria are summarised in Supplementary Table 2. The Bayesian approach quantitatively combines data from the similarly designed BIOSCIENCE and BIOSTEMI studies, which allows a more precise estimation of the clinical performance of the two study stents for the treatment of patients with STEMI. The U.S. Food and Drug Administration (FDA) has recently recognised that Bayesian analyses may lead to smaller trials while still retaining full validity of the results, by making better usage of pre-existing high-quality evidence (Guidance for the Use of Bayesian Statistics in Medical Device Clinical Trials; issued on 5 February 2010 by the FDA).
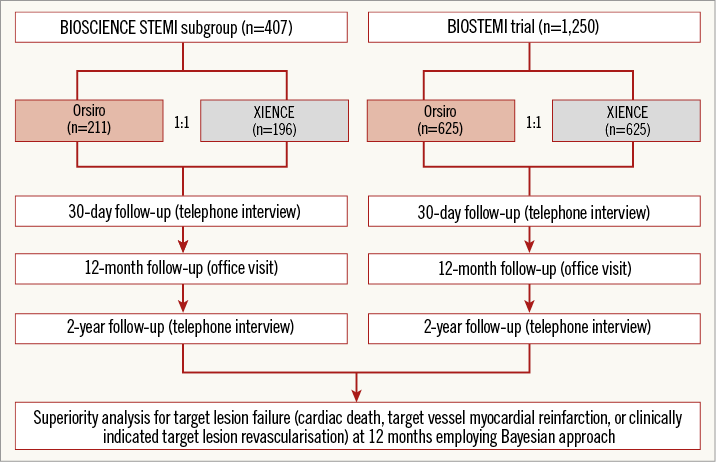
Figure 2. BIOSTEMI study design. STEMI: ST-elevation myocardial infarction
STUDY ENDPOINTS
The primary endpoint of the BIOSTEMI trial is the rate of TLF, a composite of cardiac death, target vessel reinfarction, or clinically indicated TLR, within 12 months of the index procedure. Secondary endpoints include all-cause death, cardiac death, Q-wave and non-Q-wave MI, clinically indicated and non-clinically indicated TLR, clinically indicated and non-clinically indicated TVR, target vessel failure, definite ST, and definite or probable ST at one and two years, and TLF at two years. All primary and secondary endpoint definitions are outlined in detail in the Supplementary Appendix. The study endpoints will be adjudicated by a blinded and independent clinical events committee (CEC).
STUDY POPULATION
All subjects with STEMI within 24 hours of symptom onset and who qualify for PPCI according to current guidelines1 are eligible. Subjects with coronary artery lesions suitable for DES implantation will be enrolled according to the inclusion and exclusion criteria detailed in Supplementary Table 3.
INFORMED CONSENT
All subjects will provide informed consent prior to randomisation. Considering the particular situation of subjects with STEMI requiring urgent PPCI, a specific informed consent process was developed for the purpose of the study and is detailed in the Supplementary Appendix. The study protocol, including the informed consent process, was approved by the responsible ethics committee.
RANDOMISATION
After successful crossing of the acute infarct artery target lesion, patients will be randomised in a 1:1 ratio to treatment with either BP-SES or DP-EES. Random stent allocation will be performed using an electronic web database (Cardiobase, copyright by Department of Cardiology, CTU Bern, Switzerland, and 2mt Software GmbH, Ulm, Germany) and will be stratified according to study centre, diabetes status, and presence or absence of multivessel coronary artery disease.
REVASCULARISATION PROCEDURE
PPCI will be performed according to current guidelines1. In patients with multivessel disease, revascularisation of all lesions in non-culprit vessels will be performed with the uniform use of the randomly allocated study stent within the same procedure or during subsequent staged procedures at the investigator’s discretion. Staged procedures for the treatment of non-culprit vessels are permitted within three months of the index procedure. There is no restriction with respect to type or number of lesions that are treated.
DATA COLLECTION
The schedule of measurements and follow-up plan are detailed in Supplementary Table 4. Patient data are collected in a web-based data entry system hosted at the CTU Bern. Data entry will be performed by the on-site study personnel. Central and on-site data monitoring will be organised by the CTU Bern according to a pre-specified monitoring plan. All electronic case report forms will undergo central data monitoring. On-site monitoring will be performed on the complete case report forms of the first 10 patients included at each participating site, followed by a random sample of 20% at each site. All serious adverse events will be submitted to the CTU Bern in a blinded fashion. Any death, myocardial reinfarction, revascularisation procedure, ST, cerebrovascular event, or bleeding complication will be independently adjudicated by a blinded CEC.
STATISTICAL ANALYSIS
BIOSTEMI is a superiority trial powered for the primary composite endpoint of TLF within 12 months after the index procedure. New data from patients prospectively enrolled in the BIOSTEMI trial will be combined with historical data from the subgroup of patients with STEMI included in the BIOSCIENCE trial (BIOSCIENCE STEMI) that contributed to the primary clinical endpoint analysis22,34, employing a Bayesian approach based on robust historical priors (RHP). Compared to conventional meta-analytic approaches, RHP will down-weight historical information on the primary endpoint rates, if this information turns out to be inconsistent with the new data acquired on the primary endpoint rates, for each arm separately. The analysis method is based on Schmidli et al36 and adapted to the case of a single historical trial and estimation of rates and rate ratios (RR). A detailed description of the statistical analysis is provided in the Supplementary Appendix, Supplementary Table 5-Supplementary Table 7, Supplementary Figure 1, and Supplementary Figure 2.
All patients undergoing randomisation will be included in the analysis of primary and secondary clinical outcomes in the study arm to which they were originally allocated according to the intention-to-treat principle. The primary and secondary endpoints will be analysed with Bayesian log-Poisson models applied to new data from the BIOSTEMI patients with RHP that incorporate historical information from BIOSCIENCE STEMI patients. The individual observation time of each patient will be included in these models, and incidence rates and incidence RR will be provided. The RR will be reported as the median of the posterior distribution with two-sided 95% credibility interval (CrI). Superiority of BP-SES over DP-EES will be declared if the upper limit of the CrI is ≤1.
SAMPLE SIZE
The BIOSTEMI trial is powered for superiority on the primary endpoint at 12 months using a robust Bayesian approach that incorporates historical information adopted by Schmidli et al36 and modified as detailed in the Supplementary Appendix. Power calculation was based on a Monte Carlo simulation, where information from the 407 BIOSCIENCE STEMI patients was included via RHP. Power was estimated as the number of simulated trials where superiority is declared divided by the total number of simulated trials. In BIOSCIENCE STEMI, an RR of 0.38 for BP-SES/DP-EES with respect to the primary endpoint TLF was observed at 12 months24,36. To be conservative, a less pronounced RR of 0.60, with an incidence rate for the primary endpoint of 4.2% in the BP-SES and 7.0% in the DP-EES arm, respectively, will be used for sample size calculation in the BIOSTEMI trial. We assumed a dropout rate of 5% at 12 months. With a 1:1 allocation ratio and a two-sided α=0.05, we found that enrolment of a total of 1,250 patients (625 per treatment arm) in the BIOSTEMI trial would provide more than 80% power to detect superiority of BP-SES over DP-EES.
PRE-SPECIFIED ANALYSES
Stratified analyses of the primary endpoint across major subgroups based on the BIOSTEMI subjects will be performed. Subgroup analyses of the primary endpoint will be performed with respect to diabetes, gender, age ≥65 years, BMI ≥30 kg/m2, multivessel disease, and chronic renal failure. Rates of cerebrovascular events and bleeding complications will be analysed according to the type and duration of the anticoagulant and antiplatelet strategies, as detailed in the Supplementary Appendix.
Conclusions
BIOSTEMI is a prospective, multicentre, assessor-blind, randomised, controlled trial assessing the superiority of ultrathin-strut BP-SES over thin-strut DP-EES with respect to the primary endpoint of TLF in patients with STEMI undergoing PPCI. BIOSTEMI is the first randomised controlled clinical trial comparing ultrathin-strut BP-DES with thin-strut DP-DES for PPCI.
| Impact on daily practice The optimal DES therapy for PPCI in patients with STEMI remains unclear. A novel ultrathin-strut BP-SES may improve clinical outcomes compared with the current best-in-class newer-generation DP-EES among patients with STEMI, thus potentially advancing current management strategies for PPCI. The results of the BIOSTEMI trial may entail important clinical implications for the future management of patients with STEMI undergoing PPCI in clinical practice. |
Acknowledgements
The clinical trial operational management is performed by the Clinical Trials Unit Bern, Institute of Social and Preventive Medicine, University of Bern, Bern, Switzerland.
Funding
BIOSTEMI is an investigator-initiated trial supported by a dedicated research grant from Biotronik, Bülach, Switzerland. The authors are solely responsible for the design and conduct of this study and all study analyses. The funding source was not involved in the design of the study, is not involved in data collection and management, and has and will have no role in the analysis or interpretation of the study data.
Conflict of interest statement
J.F. Iglesias reports receiving research grants to the institution from AstraZeneca and Biotronik, educational grants to the institution from Philips Volcano, and honoraria/speaker fees from AstraZeneca, Biotronik, Medtronic, Philips Volcano and Terumo. O. Muller reports receiving research grants to the institution from AstraZeneca and Biotronik, and honoraria/speaker fees from Medtronic and St. Jude Medical/Abbott. M. Roffi reports receiving institutional research grants from Abbott Vascular, Medtronic, Boston Scientific, Terumo, and Biotronik. P. Jüni reports receiving research grants to the institution from AstraZeneca, Biotronik, Biosensors International, Eli Lilly and The Medicines Company, and serves as an unpaid member of the steering group of trials funded by AstraZeneca, Biotronik, Biosensors, St. Jude Medical and The Medicines Company. M. Valgimigli reports receiving grants from AstraZeneca, and personal fees from Abbott, Amgen and Bayer, outside the submitted work. S. Windecker reports receiving grants to the institution from Biotronik, Boston Scientific, Bracco Pharmaceutical, Edwards Lifesciences, Medtronic, Terumo, and St. Jude Medical. T. Pilgrim reports receiving research grants to the institution from Biotronik, Edwards Lifesciences and Symetis, speaker fees from Boston Scientific, and reimbursement for travel from St. Jude Medical. The other authors have no conflicts of interest to declare.
Supplementary data
Supplementary Appendix. Study organisation, study design, inclusion and exclusion criteria, study endpoint definitions.
Supplementary Table 1. Study organisation.
Supplementary Table 2. Study design of trials contributing to the Bayesian analysis.
Supplementary Table 3. Inclusion and exclusion criteria.
Supplementary Table 4. Schedule of measurements and follow-up plan.
Supplementary Table 5. Comparison of rate ratios and CIs obtained by frequentist and Bayesian methods with the vague prior given in eq.3.
Supplementary Table 6. Gaussian summary of the posterior log-rate (β) of TLF obtained from the historical data.
Supplementary Table 7. Type I error and power obtained by Monte Carlo simulation under several scenarios for the event rate expected in the new BIOSTEMI trial and for values of the weighting parameter.
Supplementary Figure 1. Graphical description of how the robust historical priors are constructed.
Supplementary Figure 2. Graphical description of how the primary endpoint and secondary endpoints will be analysed.
To read the full content of this article, please download the PDF.
