Abstract
Aims: Compared with bare metal stents (BMS), early generation drug-eluting stents (DES) reduce the risk of revascularisation in patients with ST-elevation myocardial infarction (STEMI) at the expense of an increased risk of very late stent thrombosis (ST). Durable polymer coatings for controlled drug release have been identified as a potential trigger for these late adverse events and this has led to the development of newer generation DES with durable and biodegradable polymer surface coatings with improved biocompatibility. In a recent all-comers trial, biolimus-eluting stents with a biodegradable polymer surface coating were found to reduce the risk of very late ST by 80% compared with sirolimus-eluting stents with durable polymer, which also translated into a lower risk of cardiac death and myocardial infarction (MI) beyond one year.
Methods and results: The multicentre COMFORTABLE AMI trial (NCT00962416) randomly assigned 1,161 patients to treatment with biolimus-eluting stents with biodegrable polymer and bare metal stents of otherwise identical design at 11 international sites. The primary endpoint is a composite of cardiac death, target-vessel MI and target lesion revascularisation at one year. Assuming a relative risk reduction of 40% in event rates of the primary endpoint in favour of biolimus-eluting stents with biodegradable polymer, 1,064 patients will provide 80% power to demonstrate superiority. Clinical follow-up will be continued through five years.
Conclusions: The COMFORTABLE AMI trial will determine whether biolimus-eluting stents with biodegradable polymer are superior to bare metal stents of otherwise identical design. This is the first randomised controlled trial (RCT) investigating DES with a biodegradable polymer surface coating for drug release in the treatment of patients with STEMI.
Rationale
Early generation drug-eluting stents (DES), namely sirolimus-eluting stents (SES) and paclitaxel-eluting stents (PES) have been compared with bare metal stents (BMS) in the setting of ST-elevation MI (STEMI) in several randomised controlled trials enrolling more than 7,000 patients to date. A reduction in major adverse cardiovascular events (MACE) was consistently observed and mainly related to a lower risk of repeat revascularisation procedures1-7. Brar and colleagues compared outcomes between early generation DES and BMS8 in a meta-analysis of 7,352 STEMI patients and reported a 56% lower risk of repeat revascularisation in favour of DES without differences in the risk of death, MI and ST. Notwithstanding, there remains a nagging concern regarding the safety of DES in STEMI patients particularly during long-term follow-up. Histopathologic analysis of autopsy specimens revealed more inflammation, fibrin deposition and uncovered struts among lesions treated with early generation DES in patients with acute MI compared to those with stable lesions, suggesting a differential healing response depending on underlying plaque morphology9,10. Similar observations were obtained in intravascular imaging studies10,11. Late acquired stent malapposition – a potential trigger of very late stent thrombosis12,13 – was more common among DES than BMS in STEMI patients in the IVUS substudy of the MISSION study and HORIZON-AMI14,15 trial. The notion of a differential healing response between DES and BMS in STEMI patients was further substantiated by the OCT substudy of the HORIZON-AMI trial, which observed a higher rate of uncovered and malapposed struts among DES treated lesions at 13 months – a pattern much like the one observed in autopsy reports16.
A recent meta-analysis17 including 7,867 STEMI patients compared clinical outcomes between early generation DES and BMS during long-term follow-up and observed an important treatment effect over time. Overall outcomes showed improved efficacy with DES whereas safety parameters were similar. Of note, during long-term follow-up beyond one year, the risk of definite or probable stent thrombosis (ST) was significantly higher with early generation DES with a trend towards an increased risk of MI. In view of the late harm associated with early generation DES implanted among patients with STEMI, uncertainty continues to prevail regarding the optimal use of DES in this clinical setting18. While previous studies among patients with STEMI used early generation DES, newer generation devices with drug release from durable or biodegradable polymer surface coatings may provide the basis for improved biocompatibility and vascular healing19. As for DES using durable polymers, an everolimus-eluting stent (XIENCE®; Abbott Vascular, Redwood City, CA, USA) has been tested against a bare metal stent of otherwise identical design in the EXAMINATION trial. The primary endpoint of this study was the composite of death, MI and revascularisation, which was assessed at one year20.
DES with biodegradable polymer coating provide controlled drug release with subsequent degradation of the polymer material, thereby eliminating the inflammatory stimulus potentially responsible for late adverse events. Biolimus-eluting stents were designed with a biodegradable polymer (polylactic acid) applied to the stent’s abluminal surface, which is metabolised to water and carbon dioxide within six to nine months21,22. Biolimus is a highly lipophilic sirolimus analogue and inhibits proliferation of smoothmuscle cells by inducing reversible cell cycle arrest at gap 0 (G0) with similar potency to sirolimus.
Biolimus-eluting stents with biodegradable polymer (BioMatrix®; Biosensors Inc, Morges, Switzerland) have been directly compared with sirolimus-eluting stents with durable polymer (Cypher® SELECT; Cordis, Miami Lakes, FL, USA) in the large scale, randomised LEADERS trial. Non-inferiority of biolimus-eluting stents was established through four years of follow-up. During the late time period (1-4 years), the use of biodegradable polymer based biolimus-eluting stents resulted in a 80% relative risk reduction of very late definite ST, which was associated with a significant reduction in ischaemic events (cardiac death or MI) between one and four years23. Of note, a stratified analysis according to pre-specified patient characteristics suggested a pronounced reduction in MACE among the subgroup of patients presenting with STEMI at baseline (Figure 1). The improved outcome of a device with biodegradable as opposed to durable polymer surface coating in lesions with ruptured plaques further supports the notion of the improved biocompatibility of biolimus-eluting stents during long-term follow-up. As the results of a stratified analysis are only hypothesis-generating, we designed a randomised controlled trial comparing biolimus-eluting stents with biodegradable polymer with bare metal stents of otherwise identical design (Gazelle®; Biosensors Inc, Morges, Switzerland) in order to validate the safety and efficacy in the high-risk subset of patients with STEMI (COMFORTABLE AMI trial).

Figure 1. Stratified analysis of the primary endpoint MACE in the LEADERS trial at four years23. Reprinted from The Lancet, Vol. 378, with kind permission from Elsevier.
The synthesis of available data suggests that biolimus-eluting stents with biodegradable polymer potentially combine the advantages of drug-eluting stents compared with bare metal stents during the first year after implantation – namely the well-known inhibition of neointimal hyperplasia and an early antithrombogenic effect as proposed in in vitro investigations by Kolandaivelu and colleagues24. Upon biodegradation of the polymer during the time period beyond one year, biolimus-eluting stents with biodegradable polymer should show a performance more like bare metal stents and therefore prevent late adverse events previously linked to durable polymers.
In addition to advances in stent design, progress has been achieved in terms of interventional procedure and adjuvant antiplatelet and antithrombotic therapy. Namely, the systematic use of thrombus aspiration prior to stent implantation has improved outcomes following primary PCI25. Moreover, high dose clopidogrel pre-treatment led to a 42% relative risk reduction of definite ST in the CURRENT-OASIS 726 study. Novel oral P2Y12 inhibitors such as prasugrel27 and ticagrelor28 have been shown to reduce the risk of cardiac death, MI and stroke, as well as the risk of stent thrombosis compared with clopidogrel among patients with STEMI. Therefore, it remains to be determined whether newer generation DES in combination with the above-mentioned novel therapeutic strategies may further improve outcomes vis-à-vis bare metal stents.
Design
Study design and primary hypothesis
The COMFORTABLE AMI trial is a randomised, assessor blind, international, multicentre trial comparing biolimus-eluting stents with biodegradable polymer with bare metal stents of otherwise identical design implanted into culprit lesions of patients with STEMI. The trial was designed and the protocol written by the steering committee (LR, PJ, SW), and managed by the Clinical Trials Unit Bern, Switzerland. The study is powered to examine the primary hypothesis that a biolimus-eluting stent with biodegradable polymer is superior to a bare metal stent in the setting of STEMI in terms of major adverse cardiac events (MACE) – a composite endpoint of cardiac death, target vessel MI and target lesion revascularisation at one year.
Statistical analysis
On the basis of results from the HORIZON and LEADERS trials (subgroup analysis in STEMI patients), we assumed an incidence of MACE (the primary endpoint) of 14% up to one year in the bare metal stent group with a relative risk reduction of 0.60 in favour of biolimus-eluting stents. Enrolment of 1,064 patients will provide 80% power to detect a relative risk of 0.60 at a two-sided alpha of 0.05. All enrolled patients will be included in the analysis of primary and secondary clinical outcomes according to the intention-to-treat principle. We will use a Cox proportional hazards model to compare clinical outcomes between the groups.
Study population
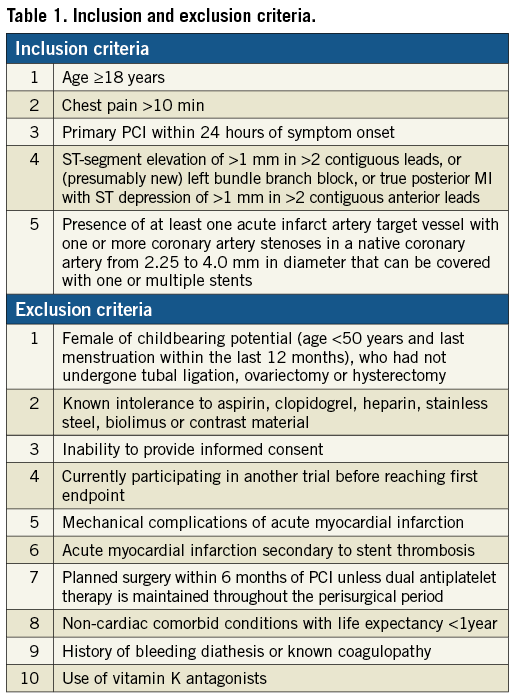
Patients aged 18 years or older with ST-elevations in at least two contiguous leads or new left bundle branch block presenting within 24 hours of onset of pain are eligible if a culprit lesion is identified during coronary angiography. Selection criteria are broad, reflecting routine clinical practice (Table 1).
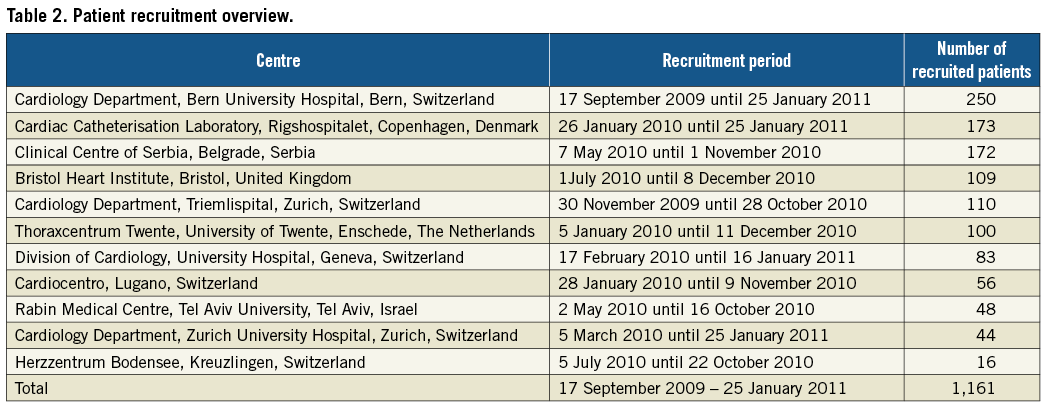
All patients will provide a first written consent prior to the randomisation procedure following oral information, whereas a second written informed consent will be obtained with the patient in a stable enough condition to read and understand the study-specific patient information. Between 19 September 2009 and 25 January 2010 (16 months), a total of 1,161 patients were randomly assigned to treatment with biolimus-eluting stents with biodegradable polymer or bare metal stents at 11 sites in Europe and Israel: Switzerland (five), Denmark (one), Israel (one), Serbia (one), The Netherlands (one), United Kingdom (one). Individual sites and the respective recruitment numbers are shown in Table 2. A total of 2,575 patients presented with ST-elevation MI during the entire inclusion period at 11 study sites, resulting in inclusion of 45.1% of patients in the COMFORTABLE AMI trial. The number of patients with exclusion criteria (e.g., patients with mechanical ventilation, unable to provide informed consent) as compared to eligible patients who did not agree to participate was not assessed. The randomisation procedure was facilitated by an electronic web database “Cardiobase” (Copyright by Cardiology Department, CTU Bern, Switzerland and 2mT software GmbH, Ulm, Germany). The allocation sequence was computer-generated and stratified according to centre.
Procedure
After visualisation of the culprit lesion, thrombus aspiration was recommended in all patients whenever aspiration was deemed technically feasible. Predilatation of the culprit lesion was left to the discretion of the operator. The randomly allocated stent had to be implanted in the culprit lesion of the target vessel and in all non-culprit lesions of the target vessel. As the superiority of DES over BMS has been convincingly established for the treatment of stable lesions, the use of a biolimus-eluting stent with biodegradable polymer was mandatory among all patients with multivessel disease requiring revascularisation of non-culprit vessels. It was advised to perform staged procedures within the first three months. At selected sites (Bern, Copenhagen, Geneva, Lugano, Zurich), intravascular ultrasound and optical coherence tomography of the culprit lesion after successful stent implantation with an angiographically satisfying result was performed as part of an imaging substudy (IBIS-4, NCT00962416).
Study drugs
The routine use of prasugrel (loading and maintenance) was recommended as a standard P2Y12 inhibitor. If prasugrel was not available, clopidogrel could be used as an alternative antiplatelet drug. Prior to the procedure, aspirin at a dose of at least 300 mg and a loading dose of 60 mg prasugrel (including patients preloaded with clopidogrel) was mandatory. If prasugrel was not available or contraindicated, clopidogrel was administered at a loading dose of 600 mg. During the procedure, unfractionated heparin was given at a dose of at least 5000 IE or 70-100 IE/Kg to maintain an activated clotting time (ACT) >250 seconds, or alternatively bivalirudin. The use of glycoprotein IIb/IIIa inhibitors was left to the discretion of the operator. After the procedure, it was recommended to administer prasugrel at a dose of 10 mg per day for at least 12 months. If prasugrel was not available or contraindicated, clopidogrel was given at a dose of 75 mg twice daily for seven days followed by a maintenance dose of 75 mg once daily (1-0-0) for at least 12 months. Following successful primary PCI, it was recommended to provide antithrombotic treatment according to local practice.
Pre-specified analyses
Stratified analysis across major clinical subgroups will be performed using multivariate logistic regression or Cox proportional hazards models. Subgroup analyses of the primary and major secondary endpoints will be performed with respect to age, gender, diabetes, renal insufficiency, lesion and stent characteristics including ostial/bifurcation lesions, differences in antiplatelet treatment strategies, SYNTAX score pre- and post-wiring of the target vessel, radial versus femoral access, biomarkers, lipid profile, and patients with recurrent instable angina without evident disease progression. Rates of bleeding and bleeding severity will be analysed according to antiplatelet strategy (clopidogrel versus prasugrel).
Substudies
A total of 103 patients at five centres (Bern, Copenhagen, Geneva, Lugano, Zurich) underwent intracoronary imaging with the use of gray-scale intravascular ultrasound, virtual histology IVUS and frequency domain optical coherence tomography (FD-OCT) of the culprit lesion immediately after stent implantation and at 13-month follow-up. The aim is to assess the stent vessel wall interactions in terms of volumetric measures such as lumen, stent, vessel area, neointimal area, apposition, coverage, plaque volume and composition behind the struts and at the stent edge. All patients undergoing intracoronary imaging of the culprit lesion will undergo quantitative coronary analysis in order to assess reference and minimal lumen diameter, percent diameter stenosis, as well as late lumen loss.
Definitions
MACE is defined as the composite of cardiac death, MI of the target vessel, and target lesion revascularisation. Myocardial infarction was defined according to the extended historical definition29 and definite, probable and possible stent thrombosis according to the Academic Research Consortium30. A more detailed outline of the definition applied in the COMFORTABLE AMI trial is provided in the appendix. All endpoints will be adjudicated by an independent event adjudication committee.
Data collection
All patient data are collected and entered into study-specific electronic case report forms by the study personnel on site. Central and on-site data monitoring is performed by the Clinical Trials Unit Bern, Switzerland. In the participating centres in Serbia and Israel, a contract monitoring company performed the monitoring according to a pre-specified monitoring plan. All electronic case record forms underwent central monitoring. Complete case record forms of the first 10 patients followed by a random sample of 10% of the remaining patient number at each centre underwent on-site monitoring. Preliminary and definite written consent were verified in 100% of cases. Patient files of 25% of patients were completely monitored with respect to event reporting. The database was held and supplied by the Clinical Trials Unit Bern, Switzerland.
Conclusion
The COMFORTABLE AMI trial is a large scale, randomised, assessor blind, multicentre, international study with a total of 1,161 enrolled patients, which will determine whether biolimus-eluting stents with biodegradable polymer are superior to bare metal stents of otherwise identical design. This is the first RCT investigating DES with biodegradable polymer surface coating for drug release in the treatment of patients with STEMI.
Acknowledgements
We gratefully acknowledge the invaluable support and contribution of the data and safety monitoring board (Jonathan Sterne, Michel Bertrand, Phillip Urban), the clinical event adjudication committee (Pascal Vranckx, Gerrit Hellige, Igal Moarof), and the lead research nurse (Therese Fahrni).
Funding
The study is supported by an unrestricted grant from Biosensors SA, Morges, Switzerland, and a grant from the Swiss National Science Foundation (Grant 33CM30-124112), Bern, Switzerland to SW and PJ. LR is the recipient of a research fellowship (SPUM) funded by the Swiss National Science Foundation. The funding sources were not involved in the study at any point in time. Specifically, there was no involvement in the design of the study, site selection, data collection, and funding source will not be involved in the analysis or interpretation of the data or in manuscript writing.
Conflict of interest statement
CTU Bern, which is part of the University of Bern, has a staff policy of not accepting honoraria or consultancy fees. BM has received research grants to the institution from Abbott and Cordis. SW has received research contracts to the institution from Abbott, Boston Scientific, Biosensors, Cordis, and Medtronic. PJ is an unpaid steering committee or statistical executive committee member of trials funded by Abbott Vascular, Biosensors, Medtronic, and Johnson and Johnson. All other authors have no conflict of interest to declare.
Appendix
Study organisation
Sponsor: Clinical Trials Unit Bern, Bern, Switzerland and Cardiology Department, Bern University Hospital, Bern, Switzerland
Funding: Unrestricted grant from the Swiss National Science Foundation (Grant 33CM30-124112), Bern, Switzerland; Biosensors Europe S.A., Morges, Switzerland
Steering committee: Lorenz Räber, Peter Jüni, Stephan Windecker
Clinical adjudication committee: Pascal Vranckx, Hasselt, Belgium (Chair); Gerrit Hellige, Aarau, Switzerland; Igal Moarof, Aarau, Switzerland
Data coordination and analysis: Clinical Trials Unit Bern, Bern University Hospital, Switzerland (Dik Heg, Sven Trelle, Peter Jüni)
Site management and on-site data monitoring: Clinical Trials Unit Bern, Bern, Switzerland (Brigitte Wanner, Anna Plym, Lucia Kacina); Focus, Belgrade, Serbia (Dragana Trifunovic); Ben Gurion University, Israel Monitor Tel Aviv (Leonid Kalichman), Tel Aviv, Israel
Central monitoring: Clinical Trials Unit Bern, Bern University Hospital, Bern, Switzerland (Sandro Baumgartner, Timon Spörri)
Data safety and monitoring board: Jonathan Sterne, Bristol, United Kingdom (chair); Michel E. Bertrand, Lille, France; Phillipe Urban, Geneva, Switzerland
Qualitative and quantitative coronary angiographic core laboratory: Cardiology Department, Bern University Hospital, Bern, Switzerland (Lorenz Räber, Masanori Tanikawi)
Intravascular ultrasound core laboratory analysis: Cardialysis BV, Rotterdam, The Netherlands (HM Garcia-Garcia)
Optical coherence tomography analysis: Cardialysis B.V., Rotterdam, The Netherlands (HM Garcia-Garcia)
Secondary endpoints
All cause death; death or MI; cardiac death or MI; any MI (Q-wave and non-Q-wave); target vessel failure (composite of cardiac death, target vessel MI, target vessel revascularisation); composite of death or MI or target vessel revascularisation; target vessel revascularisation; any revascularisation; Academic Research Consortium definite, probable and possible stent thrombosis; repeat hospitalisation due to angina. Additional endpoints that are not directly related to the stent comparison include: transient ischaemic attacks; stroke; bleeding according to Thrombolysis In Myocardial Infarction (TIMI), Global Use of Strategies to Open Occluded Arteries (GUSTO) and Bleeding Academic Research Consensus (BARC); renal insufficiency according to Risk, Injury, Failure, Loss, and End-stage kidney disease (RIFLE); access site complications.
Endpoint definitions
1. Death
Cardiac death: All deaths are considered cardiac unless an unequivocal non-cardiac cause can be established.
Vascular death: Death caused by non-coronary vascular causes, such as cerebrovascular disease, pulmonary embolism, ruptured aortic aneurysm, dissecting aneurysm, or other vascular cause.
Non-cardiovascular death: Any death not covered by the above definitions, including death due to infection, sepsis, pulmonary causes, accident, suicide or trauma.
2. Myocardial infarction
Target vessel myocardial infarction is any infarction that is not clearly attributable to a non-target vessel.
2.1.1 Spontaneous Q-wave MI (>48 hours after index intervention)
The diagnosis of Q-wave myocardial infarction requires new pathological Q-waves in two or more contiguous ECG leads. Elevated cardiac biomarkers or a history of chest pain or other acute symptoms consistent with myocardial ischaemia are supportive of the diagnosis and should be documented but not needed to make the diagnosis (silent myocardial infarction).
2.1.2 Spontaneous non-Q-wave MI (>48 hours after index intervention)
Appropriate cardiac enzyme data (respecting top down hierarchy): 1) CKMB >1*URL, or 2) in the absence of CKMB, standard troponin >1*URL (high sensitivity troponin is considered as missing troponin), or 3) in the absence of troponin and CKMB: CEC decision upon clinical scenario and with guidance of high sensitivity troponin. The appropriate enzyme data should be considered in conjunction with at least one of the following parameters: 1) evidence of myocardial ischaemia as recognised by clinical symptoms, or 2) evidence of myocardial ischaemia as recognised by electrocardiographic changes, or 3) evidence of myocardial ischaemia as recognised by cardiac imaging of new loss of viable myocardium or new regional wall motion abnormality.
2.2.1 Baseline biomarkers of myocardial damage: CK and/or CK-MB >1xURL or acute MI in progress
Scenario A) CK (or CK-MB) from index MI has not yet reached its maximum level: recurrent thoracic chest pain or ischaemia equivalent >20 minutes (or new ECG changes consistent with MI) and appropriate cardiac enzyme data: 1) a rise in CK within 24 hours of the index event >2*URL (confirmed by either CK-MB or troponin >1*URL) and ≥50% above the previous level, or 2) in the absence of CK: a (post-PCI) rise in CK-MB within 24 hours of the index event >3*URL and ≥50% above the previous level, or 3) in the absence of CK and CK-MB: a (post-PCI) rise of troponin within 24 hours of the index event >3*URL and ≥50% above the previous level.
Scenario B) Elevated CK (or CK-MB) following the index MI has peaked and CK level has returned
Scenario C) CK (or CK-MB) following the index MI has peaked and CK level has not returned
2.2.2 Baseline biomarkers of myocardial damage (CK and CK-MB and trop< 1*URL) and no acute MI in progress
Scenario A) New pathologic Q-waves in ≥2 contiguous ECG leads and: 1) any CK-MB >1*URL, or 2) in the absence of CK-MB: troponin >1*URL, or 3) in the absence of CK-MB and troponin: CK >1*URL, or 3) in the absence of CK-MB and troponin and CK: CEC decision upon clinical scenario.
Scenario B) Appropriate cardiac enzyme data: 1) CK ≥2* URL confirmed by either CK-MB >1*URL, or 2) in the absence of CK-MB: troponin >1*URL, or 3) in the absence of CK-MB and troponin: CEC decision upon clinical scenario. In the absence of CK: CK-MB >3*URL or in the absence of CK and CK-MB: troponin >3*URL.
Target vessel myocardial infarction
Any myocardial infarction not clearly attributable to a non-target vessel.
Target vessel (TV)
The TV is defined as the index major coronary artery (LAD, LCX or RCA and its side branches), which was in physical contact with any component (guiding catheter, guidewire, balloon catheter, etc.) of the angioplasty hardware during the initial procedure.
Target lesion (TL)
The target lesion is the treated lesion starting 5 mm proximal of the stented lesion and ending 5 mm distal of the stented lesion.
Target lesion revascularisation (TLR)
TLR is defined as any repeat percutaneous intervention of the target lesion or bypass surgery of the target vessel. All TLR will be recorded in the CRF (re-intervention justification form) and be classified prospectively as justified or non-justified by the investigator prior to the re-intervention as well as retrospectively by the independent angiographic core laboratory (in the case of percent diameter stenosis).
Clinically indicated revascularisation
Angiography at follow-up shows a percent diameter stenosis ≥50% (quantitative coronary angiography [QCA]) and if one of the following occurs: a positive history of recurrent angina pectoris presumably related to the target vessel; or objective signs of ischaemia at rest or during exercise tests presumably related to the target vessel; or abnormal results of any invasive functional diagnostic test. A TLR with a diameter stenosis ≥70% (QCA) in the absence of the above-mentioned ischaemic signs or symptoms is also considered justified. On the contrary, non-clinically indicated TLR are interventions for all stenoses <50% in the presence or absence of ischaemic signs or symptoms or all stenoses ≥50% but <70% without ischaemic signs or symptoms.
Device success
Achievement of a final diameter stenosis of <50% as measured by quantitative coronary angiography (QCA) during the initial procedure with the use of the study stent only.
Lesion success
Achievement of a final diameter stenosis of <50% as measured by quantitative coronary angiography (QCA) during the initial procedure with the use of any device.
Procedural success
Clinical devices success (<50% post-procedural stenosis with study stent) together with the absence of any in-hospital major adverse cardiac events.
Stent thrombosis
Definite, probable and possible stent thrombosis is defined according to the Academic Research Consortium.
Transient ischaemic attack
Rapidly developed clinical signs of global disturbance of cerebral function lasting <24 hours, regardless of the presence of an acute clinically relevant brain lesion in imaging.
Ischaemic stroke
Rapidly developed clinical signs of focal or global disturbance of cerebral function lasting >24 hours with imaging of an acute clinically relevant brain lesion. Ischaemic cerebral infarctions with conversion to haemorrhage are listed here. The diagnosis of a intracerebral haemorrhage must be confirmed by cerebral imaging.
Bleeding
Bleeding will be defined according to the TIMI31, GUSTO32, and BARC33 criteria as defined elsewhere.