
Abstract
Aims: The BIOFLOW-IV clinical trial was designed for regulatory submission in Japan. It assessed the safety and efficacy of a new third-generation sirolimus-eluting stent system with bioresorbable polymer (Orsiro, BP-SES) compared with an everolimus-eluting stent system with permanent polymer (XIENCE Prime/Xpedition, PP-EES).
Methods and results: This prospective, international, multicentre, 2:1 randomised, non-inferiority trial enrolled 575 patients (385 BP-SES and 190 PP-EES) with 659 stenotic de novo lesions. Of these, 137 patients (23.8%) were Japanese. Follow-up until five years is ongoing. We herein report outcomes at 12 months. Baseline parameters were well balanced. Device success was 98.9% for BP-SES versus 99.6% for PP-EES, p=0.670. Non-inferiority related to 12-month target vessel failure was met (pnon-inferiority <0.001). Further, there was no significant difference in clinical outcomes between the groups. The target vessel failure rate was 5.5% for BP-SES and 7.5% for PP-EES, the target lesion failure rate was 4.2% versus 5.4%, and the definite or probable stent thrombosis rate was 0.8% versus 0%.
Conclusions: The randomised BIOFLOW-IV trial provides further evidence on the safety and efficacy of the Orsiro BP-SES and its non-inferiority to the current benchmark, an everolimus-eluting permanent polymer stent. ClinicalTrials.gov: NCT01939249
Introduction
First-generation drug-eluting stents (DES) led to a reduction of restenosis compared to bare metal stents, but at the cost of elevated stent thrombosis rates. The annual rate of definite very late stent thrombosis was 0.6-0.7% and the major adverse cardiac events rate increased constantly by 2.6% per year1. Second-generation DES with thinner struts and an improved polymer-drug combination improved results, particularly of very late stent thrombosis2,3. The Orsiro bioresorbable polymer sirolimus-eluting stent (BP-SES) (Biotronik AG, Bülach, Switzerland) is a third-generation DES which is based on an ultra-thin cobalt-chromium stent. According to a recent state-of-the-art paper, it currently has the thinnest struts of marketed DES4. It has a unique hybrid coating consisting of a bioresorbable drug-polymer combination which –after bioresorption– only leaves the bare metal stent in the vessel, which is covered by a passive coating layer of amorphous silicon carbide5.
Numerous studies have been conducted with this novel device with more than 30,000 patients enrolled so far; however, enrolment was predominantly in Europe. The aim of the randomised BIOFLOW-IV trial was to provide further evidence on the safety and efficacy of the device, including Japanese centres where BP-SES was an investigational device and used for the first time. From the study initiation onwards, the study was set up to meet the Japanese regulatory requirements and organised to meet the data integrity from the Good Clinical Practice conformity perspectives. Subsequently, data from BIOFLOW-IV were used as the primary data set for the Japanese market approval.
Methods
STUDY DESIGN AND RANDOMISATION
BIOFLOW-IV is a prospective, intercontinental, multicentre, randomised controlled, non-inferiority trial, comparing BP-SES with permanent polymer everolimus-eluting stents (PP-EES). The details of the study design are available at ClinicalTrials.gov (NCT01939249). Subjects were randomised in a 2:1 ratio to either the BP-SES Orsiro or the PP-EES XIENCE Prime/Xpedition (Abbott Vascular, Santa Clara, CA, USA). The randomisation was performed intraprocedurally once all inclusion and exclusion criteria were verified, using an electronic data capture system to assign patients to treatment groups, a block size of 3, and stratification by site and diabetes status. Subjects were informed about the treatment allocation after the index procedure. The trial was conducted in accordance with the Declaration of Helsinki, Japanese Good Clinical Practice, and local and national regulations. The study was monitored including 100% source document verification. A clinical events committee (CEC) adjudicated all serious adverse events and a core laboratory (MedStar Health Research Institute, Washington, DC, USA) assessed the baseline and procedural angiographic parameters. Follow-up assessments were scheduled at 1, 6, and 12 months; annual follow-up up to five years is ongoing. An office visit was only required for the 12-month follow-up visit; the remaining visits could be conducted by phone.
PARTICIPANTS
A total of 575 eligible coronary artery disease (CAD) patients were enrolled at 46 sites in Japan (n=12), Europe (n=29), Australia (n=2), and Israel (n=3). Main inclusion criteria were CAD with stenotic de novo lesions in up to two separate native coronary arteries with a reference vessel diameter ≥2.50 mm and ≤3.75 mm, lesion length ≤26 mm, and target vessel Thrombolysis In Myocardial Infarction (TIMI) flow ≥2. Main exclusion criteria were evidence of myocardial infarction (MI) within 72 hours prior to the procedure, left ventricular ejection fraction ≤30%, impaired renal function (i.e., serum creatinine >2.5 mg/dl), planned intervention of the target vessel(s) after the index procedure and planned intervention of the non-target vessel within 30 days after the index procedure, life expectancy less than 12 months, three-vessel CAD at the time of the procedure, and the target lesion being located in the left main, located or supplied by a bypass graft, involving a side branch >2.0 mm, being an ostial lesion, being heavily calcified, or containing a thrombus. Subjects were deemed eligible after they had signed the consent and all inclusion and exclusion criteria had been fulfilled.
STUDY DEVICES
The BP-SES is an ultra-thin cobalt-chromium alloy platform (60 μm struts for stent sizes with 3.0 mm diameter and below) with a hybrid coating: the passive coating with amorphous silicon carbide encapsulates the stent surface by covalently binding to the metal stent surface5,6. It reduces ion release from the metal stent platform and minimises the interaction between the stent platform and the surrounding tissue. It also reduces the release of allergenic metal ions by 96%, which accordingly leads to a lower rate of corrosion and a lower risk of tissue inflammation7,8. The active coating consists of a biodegradable poly-L-lactic acid polymer from which sirolimus is released9. PP-EES is a cobalt-chromium metal platform (81 μm struts) with a durable polymer (poly-n-butyl-methacrylate [PBMA]) which adheres to the stent and drug coating. The co-polymer consisting of vinylidine fluoride and hexafluoropropylene (PVDF-HFP) serves as drug matrix and releases everolimus1.
INTERVENTION
BP-SES was an investigational device in Japan and therefore, instead of the device name, the code BTR-1131 was used in compliance with Japanese Good Clinical Practice. Both stents were available in diameters of 2.5, 2.75, 3.0, and 3.5 mm, and PP-EES additionally in a 3.25 mm diameter. Stent lengths were 9, 13, 15, 18, 22, 26, and 30 mm for BP-SES and 8, 12, 15, 18, 23, 28, and 33 mm for PP-EES. For the mandatory predilatation, only balloon angioplasty was allowed. Multiple focal lesions were considered as a single lesion if they could be covered by one stent. Implantation technique had to follow the respective instructions for use; the stent should extend beyond the lesion by a minimum of 2 mm both proximally and distally. More than one study stent was not allowed except in case of suboptimal results or bail-out situations. Post-dilatation was allowed at the discretion of the investigator. Post intervention, acetylsalicylic acid life-long and dual antiplatelet therapy (DAPT) for at least six months was recommended.
ENDPOINTS AND DEFINITIONS
The primary endpoint was non-inferiority to PP-EES for 12-month target vessel failure (TVF), defined as a composite of cardiac death, target vessel Q-wave or non-Q-wave MI10, emergent coronary artery bypass grafting (CABG), or clinically driven target vessel revascularisation (TVR) and presented as frequency. Secondary clinical endpoints were TVF, target lesion failure (TLF), defined as a composite of cardiac death, target vessel Q-wave or non-Q-wave myocardial infarction (MI), and clinically driven target lesion revascularisation (TLR); MI10; clinically driven TLR; clinically driven TVR; all-cause mortality; cardiac and non-cardiac death; a composite of death and MI; a composite of death, MI, and TVR; rate of probable or definite stent thrombosis11, and cerebrovascular events. Device success was defined as final residual diameter stenosis <30% by quantitative coronary angiography (QCA), using the assigned device only and including successful delivery of the stent at the target lesion, appropriate stent deployment, and successful removal of the stent delivery system. Procedural success was defined as final diameter stenosis <30% by QCA using any percutaneous method, without TLF during the hospital stay.
STATISTICAL ANALYSIS
The sample size was calculated using the Equation 1 Fleiss formula and was based on the following criteria: expected 12-month BP-SES TVF frequency of 5.4%, expected PP-EES TVF frequency of 5.4%, 6.0% non-inferiority margin, one-sided significance level of 0.025, power of 0.80, and an expected withdrawal/drop-out rate of 10%. Per calculation, 555 patients were required (370 BP-SES, 185 PP-EES).
The current analysis is based on the modified intention-to-treat population, meaning that subjects were allocated to the group they were randomised to irrespective of stent placement. Subjects who were randomised but did not receive a study stent were followed up to 12 months only. The visit time window was used for respective analysis (for 12-month outcomes, the time interval up to 379 days was considered). Continuous variables are presented as mean±standard deviation (SD) and categorical variables as n (%). Chi-square, Fisher’s and Wilcoxon tests were used for comparison between the treatment groups. The secondary clinical endpoints were presented as Kaplan-Meier estimates including 95% confidence intervals, and groups were compared using the log-rank test. The statistical analyses were carried out using SAS 9.4 (SAS Institute, Cary, NC, USA).
Results
BASELINE PATIENT AND PROCEDURAL CHARACTERISTICS
From September 2013 to January 2015, 575 patients with 659 lesions were enrolled (Figure 1). Of these, the 137 Japanese patients were enrolled from September 2013 until June 2014, and the remaining non-Japanese patients from February 2014 until January 2015. Enrolment per country is displayed in Supplementary Figure 1.
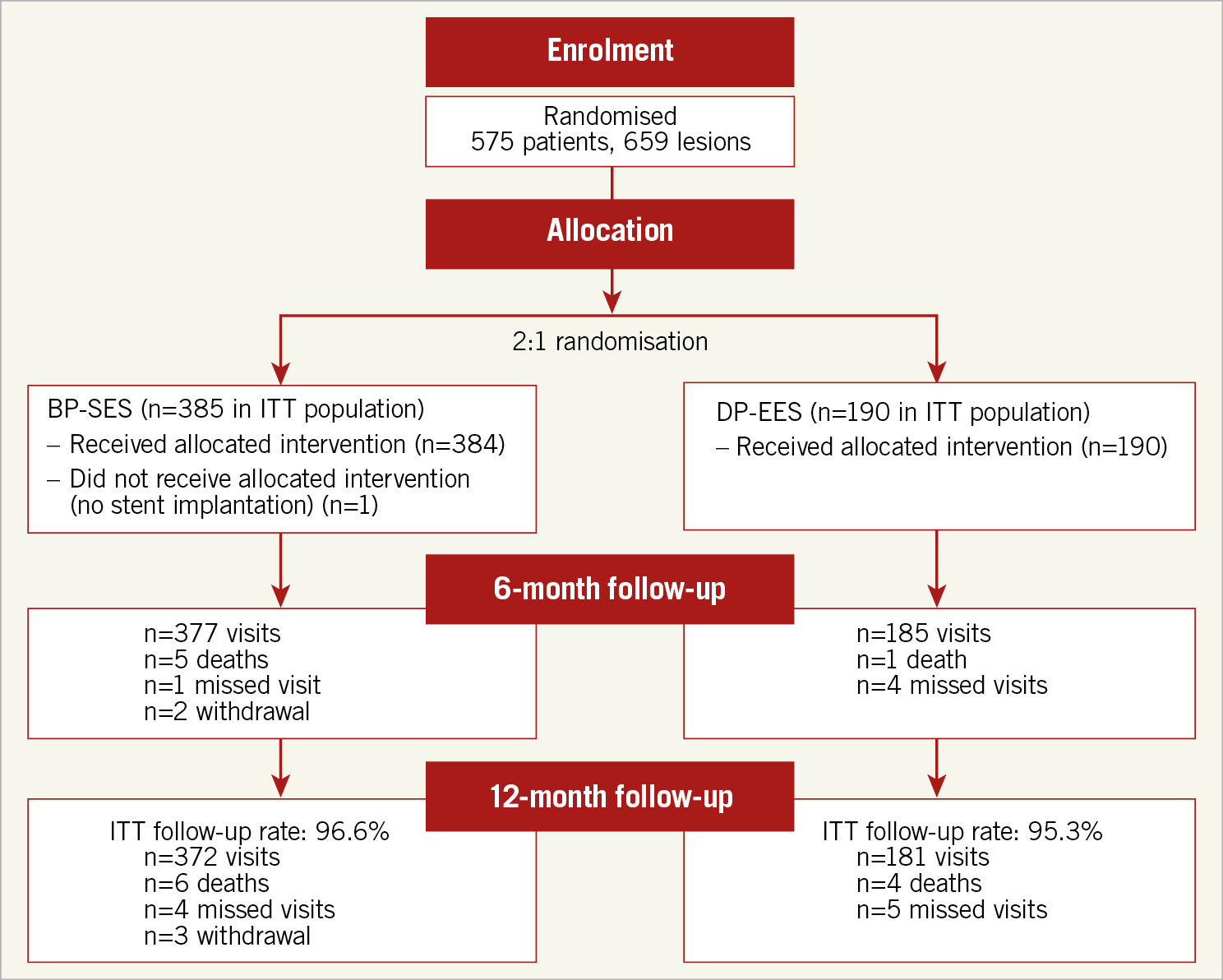
Figure 1. Patient flow chart.
Baseline parameters were well balanced amongst the BP-SES and PP-EES groups (Table 1). Mean age was 64.7±9.6 years, 74.1% were male, 30.6% had diabetes, and 30.6% a history of MI.
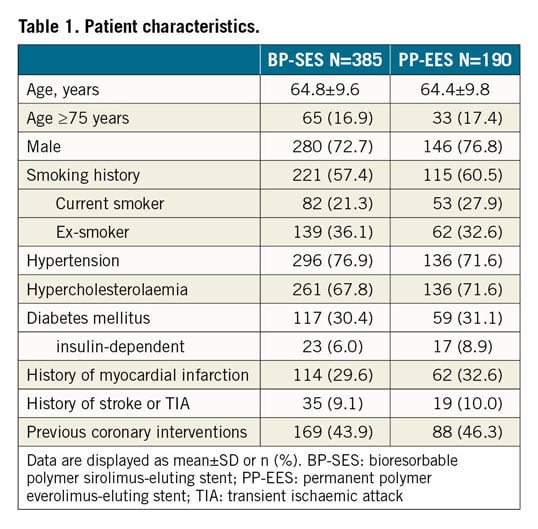
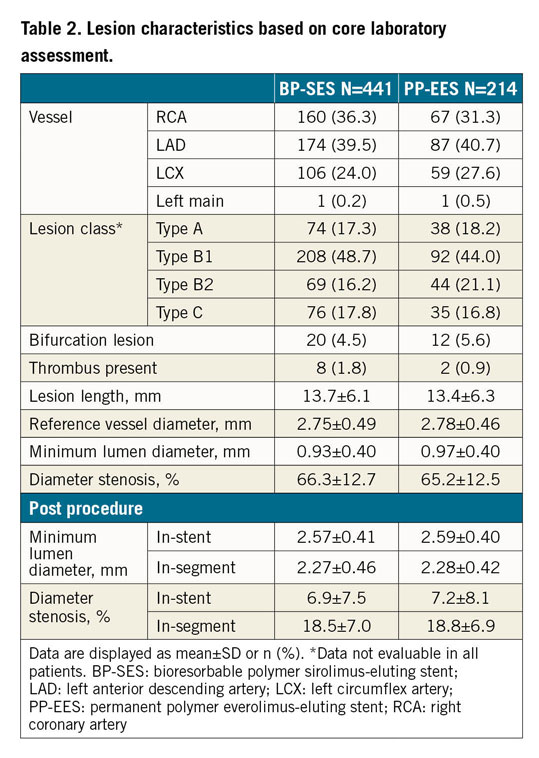
Vascular access was predominantly radial (66.3%), followed by femoral (31.3%) and brachial (2.4%) access. Predilatation was performed in 84.0% of BP-SES and 80.1% of PP-EES lesions (p=0.216) and post-dilatation in 41.3% versus 46.3% (p=0.225). Mean stent length was 18.6±5.4 mm versus 18.2±6.2 mm, and mean stent diameter was 3.03±0.38 mm versus 3.04±0.38 mm. Device success was 98.9% (467/472 stents) for BP-SES and 99.6% for PP-EES (227/228 stents) (p=0.670) and clinical procedural success based on the universal definition for MI was 96.1% (370/385) versus 95.8% (182/190) (p=0.856). Patients were discharged at 1.5±2.2 days post procedure on average, ranging from 0 to 32 days. Lesion characteristics assessed by the core laboratory indicated no significant differences between BP-SES and PP-EES; lesion details are provided in Table 2.
CARDIAC MEDICATIONS
At six months, 85.9% (BP-SES) versus 84.9% (PP-EES) of the patients were on DAPT, and, at 12 months, 59.1% (BP-SES) versus 46.4% (PP-EES) of the total population. Statin use was nearly identical in BP-SES and PP-EES patients, ranging from 83.4% to 83.9% up to 12-month follow-up.
FOLLOW-UP RATES AND CLINICAL OUTCOMES
The follow-up rate at 12 months was 96.6% in the BP-SES group and 95.3% in the DP-EES group (median follow-up duration was 367 days for all patients and 365 for those patients who did not reach 379 days).
The primary endpoint, 12-month TVF frequency using the universal definitions10, was observed in 5.1% of BP-SES patients versus 6.6% of PP-EES patients (difference –2.27% and 95% CI: –6.83, 2.29). Non-inferiority of BP-SES versus PP-EES was confirmed (p<0.001). There was no significant difference in relation to clinical outcomes. The secondary endpoints, Kaplan-Meier failure estimates at 12 months, were as follows: TVF was 5.5% for BP-SES and 7.5% for PP-EES, TLF was 4.2% versus 5.4%, and clinically driven TLR was 2.1% versus 1.8% (Figure 2, Table 3). Cardiac death was 0.0% in the BP-SES group and 0.5% in the PP-EES group. This event was one sudden death during swimming on day 202. The autopsy did not reveal any suspicion of myocardial infarction or stent thrombosis.
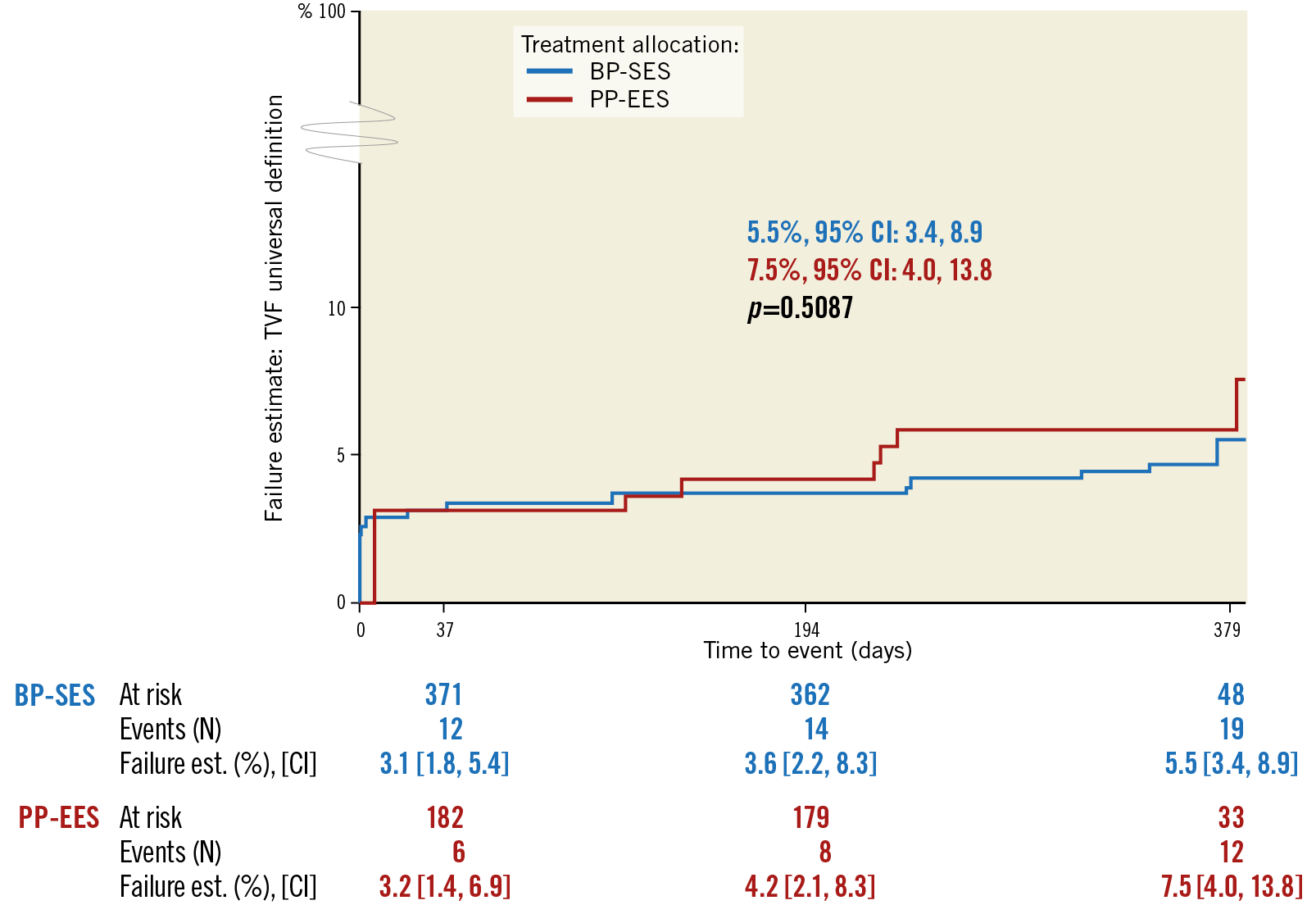
Figure 2. Kaplan-Meier failure estimates for target vessel failure in BP-SES versus PP-EES up to 12-month follow-up. CI: confidence interval; BP-SES: bioresorbable polymer sirolimus-eluting stent; PP-EES: permanent polymer everolimus-elutng stent; TVF: target vessel failure
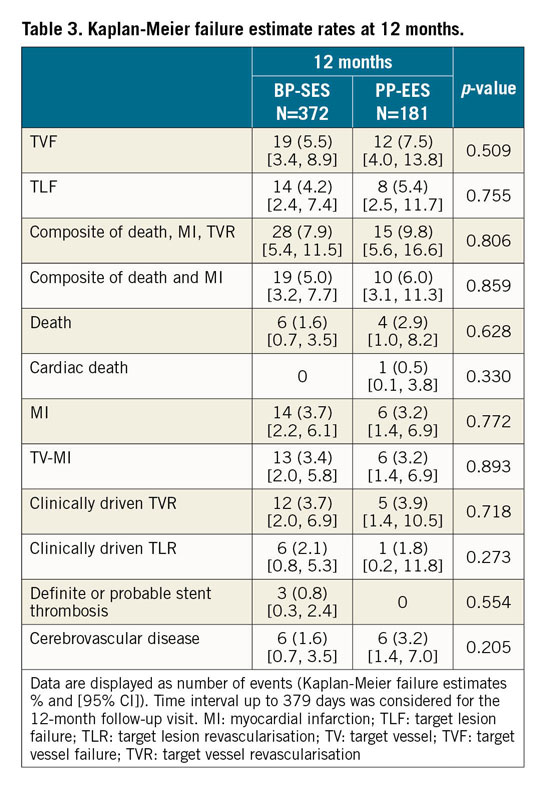
Three stent thromboses were adjudicated by the CEC in the BP-SES group (Supplementary Appendix 1). Case 1 had a heavily calcified left anterior descending artery and extensive dissection after predilatation. The stent thrombosis of case 2 was associated with high on-treatment clopidogrel resistance as measured by VerifyNow, and case 3 had continued elevation of ST and cardiac enzymes before the index procedure. The rates of definite or probable stent thrombosis were 0.8% (BP-SES) versus 0.0% (PP-EES) with no significant difference between the treatment groups (p=0.554).
REGIONAL ANALYSIS
The characteristics of the patient baseline, procedural and clinical outcomes in the Japanese versus the non-Japanese group were assessed in order to verify the applicability of intercontinental data for regulatory approval in Japan.
Japanese patients were significantly older, had more hypertension and hypercholesterolaemia, a higher incidence of prior stroke or transient ischaemic attack, more previous coronary interventions and less lesion calcification compared to non-Japanese (Supplementary Table 1). During the procedure, predilatation and post-dilatation were more frequent (98.6% and 78.8%) in Japanese subjects compared to 78.2% and 32.7% in non-Japanese subjects (p<0.001). Less pressure was applied for dilatation, and inflation time was longer in Japanese patients. Furthermore, the average stent length was longer in the Japanese cohort. During follow-up, more Japanese patients were on DAPT: 95.5% of Japanese subjects compared to 82.5% of non-Japanese subjects at six months (p<0.001) and 81.1% versus 46.8% at 12 months (p<0.001). No significant difference was observed in the clinical endpoints comparing Japanese versus non-Japanese subjects and between the BP-SES and PP-EES groups in Japanese patients (Supplementary Table 2-Supplementary Table 4, Supplementary Figure 2, Supplementary Figure 3).
Discussion
The main findings of the BIOFLOW-IV randomised trial are very good clinical outcomes in the BP-SES and the PP-EES groups without statistically significant differences between them. The primary endpoint, non-inferiority of BP-SES to PP-EES, was met with p<0.001.
The clinical outcomes of the BP-SES group are consistent with several randomised controlled trials comparing BP-SES with the PP-EES stents, mainly in a European population. They univocally report similar outcomes amongst both stents. TVF at 12 months was 5.0% compared to 4.0% in Korean patients enrolled in the ORIENT trial, 5% in BIO-RESORT, 7% in BIOFLOW-V, 8.1% in BIOSCIENCE and 9.3% in BIOFLOW-II12,13,14,15,16. TLF at 12 months was 4.2% compared to 2.4% in ORIENT, 4% in BIO-RESORT, 5.1% in BIOFLOW-III, 6% in BIOFLOW-V, 6.5% in BIOFLOW-II, and 6.7% in BIOSCIENCE12,13,14,15,16,17. Definite or probable stent thrombosis at 12 months was 0.8% compared to 0% in the ORIENT and BIOFLOW-II studies, and <1% in BIO-RESORT and BIOFLOW-V. The recent publication of the BIOFLOW-V randomised trial included not only centres from Europe, but also centres from the USA, Canada, Australia, New Zealand, South Korea and Israel, and reported superior TLF and myocardial infarction rates for BP-SES compared to PP-EES12. Aside from these trials, randomised trials also showed similar results of BP-SES compared to durable polymer zotarolimus-eluting stents13,14, and everolimus-eluting or biolimus-eluting biodegradable polymer stents13,18.
Likewise, the 12-month outcomes of BIOFLOW-IV are similar to the nine-month outcomes of the CENTURY-II randomised trial, comparing another sirolimus-eluting biodegradable polymer stent (Ultimaster®; Terumo Corp., Tokyo, Japan) with the PP-EES in 1,101 patients19. Specifically, TVF at 12 months was 5.0% for BP-SES in our series compared to 6.0% at nine months for the Ultimaster in CENTURY-II, and definite or probable stent thrombosis was 0.8% compared to 0.9%, respectively. Comparing outcomes in Japanese patients only, in our study, the TVF rate at 12 months in Japanese patients was 2.2% for BP-SES as compared to 5.0% in 100 patients of the RESOLUTE Japan trial treated with a permanent polymer zotarolimus-eluting stent20. Definite or probable stent thrombosis was absent in the latter, but a 4.0% myocardial infarction rate was observed.
Long-term follow-up is required to clarify the influence of differences in patient baseline and procedural characteristics and DAPT on TVF and stent thrombosis between Japanese and non-Japanese subjects. Even though the 12-month TLF rate is numerically higher in the non-Japanese group, this has to be interpreted carefully as the study was not designed to detect differences in clinical endpoints between Japanese and non-Japanese subjects.
Additionally, long-term follow-up observation is expected to elucidate further the potential benefits of the passive coating of the BP-SES stent9. The already available five-year clinical results of the BIOFLOW-II study21, the BIOFLOW-III registry22 and of the BIOSCIENCE study23 support the concept of reduced release of allergenic metallic ions and reduction of tissue inflammation because of the passive coating method with a 0% definite or probable stent thrombosis rate at five years in BIOFLOW-II and 0.7% in BIOFLOW-III, as well as a five-year stent thrombosis rate of 1.6% in BIOSCIENCE.
Limitations
The study has several limitations. Inclusion and exclusion criteria were restrictive, excluding patients with acute myocardial infarction for Japanese regulatory purposes. Therefore, results only apply to the specific patient population presented herein. Additionally, the study was not powered to detect differences between the Japanese and non-Japanese populations and therefore respective conclusions are speculative. The pre-defined non-inferiority margin seemed high at 6.0%; however, results clearly show that the difference in TVF rates between the treatment arms was much narrower and well within the non-inferiority margin of 6%. Investigators, the CEC and the independent core laboratory were not blinded to the treatment allocation which could have introduced a bias.
Conclusions
The randomised BIOFLOW-IV trial provides further evidence on the safety and efficacy of the BP-SES with biodegradable polymer and its non-inferiority to a current state-of-the-art everolimus-eluting permanent polymer stent. The data further indicate its safety and efficacy in the Japanese population. Long-term observation of the BIOFLOW-IV subjects is expected to assess further the potential clinical benefits of the BP-SES.
|
Impact on daily practice The ultra-thin cobalt-chromium alloy platform BP-SES, Orsiro, demonstrated similar safety and efficacy outcomes at 12 months to the current benchmark PP-EES in this study. The occurrence of stent thrombosis was low with no late stent thrombosis, supporting the safe use of the ultra-thin strut stent in daily practice. Patient baseline characteristics and procedural differences observed between Japan and Europe did not translate into differences in clinical endpoints and, ultimately, BIOFLOW-IV led to device approval in Japan. |
Acknowledgements
We would like to thank Stephanie Sauter and Barbara Widmann (employees of Biotronik AG) for their help in preparing the manuscript.
Funding
The BIOFLOW-IV study was funded by Biotronik AG, Switzerland.
Conflict of interest statement
R. Tölg declares receiving speaker’s honoraria from Biotronik. B. Witzenbichler declares having received financial support from Biotronik for study coordinator’s labour costs. M. Haude reports study grants and personal fees from Biotronik, Abbott Vascular, Cardiac Dimensions, and Philips. T. Slagboom declares having a personal consultancy agreement with Biotronik before and during the study. R. Waksman reports grants and personal fees from Abbott Vascular, AstraZeneca, Biosensors, Biotronik, Boston Scientific and Chiesi, personal fees from Amgen, Corindus, Lifetech Medical, Medtronic, Philips Volcano and Pi-Cardia Ltd, being an investor in MedAlliance, and receiving grants from Edwards Lifesciences. The other authors have no conflicts of interest to declare.
