Abstract
Aims: We report the initial human evaluation of the novel BioMimeTM sirolimus-eluting stent (SES) (Meril Life Sciences Pvt. Ltd., Gujarat, India) with an ultra-thin stent platform (65 μm) and a biodegradable polymer for the treatment of de novo coronary lesions.
Methods and results: The meriT-1 trial was a prospective, non-randomised, single-arm, single-centre, first-in-human evaluation of the safety, feasibility and performance of the BioMime SES. Lesion criteria included non-occlusive stenosis ≤19 mm in length located in native coronary vessels. Clinical follow-up (FU) was performed at 1, 8 and 12 months; all patients were assigned for angiographic FU at eight months. A total of 30 patients (30 lesions) were enrolled between March 2009 and February 2010. Mean age was 49.9 years, 30% were diabetics, and 36.7% had previous myocardial infarction (MI). Baseline median [25%, 75% interquartile range] lesion length, reference diameter and % diameter stenosis were 15.51 mm [12.74, 20.27], 2.94 mm [2.71, 3.34], and 80.5% [67.0%, 90.7%], respectively. Overall, there was one stent implanted per lesion and procedural success was 100%. At eight-month angiographic FU (26/30), median in-stent late lumen loss was 0.15 mm [0.09, 0.33]; also, there were no cases of binary restenosis within the treated segment. Clinical FU at 12 months (100%) demonstrated absence of MACE (cardiac death, MI and target lesion revascularisation) and stent thrombosis (ST).
Conclusions: The novel BioMime SES demonstrated excellent performance in single coronary lesions including high procedural success and efficacy, as demonstrated by the relatively low late lumen loss (a surrogate of neointimal hyperplasia) at eight-month angiographic FU. Overall, there were no safety concerns in this preliminary evaluation including absence of MACE or ST up to 12 months.
Introduction
The impact of strut thickness and cell design on neointimal hyperplasia (NIH) with bare metal stents (BMS) has previously been reported, given that metallic platforms with thinner struts and open cell design appeared to benefit from significant reductions in NIH as compared to platforms with thicker struts and closed cell design1-4. Drug-eluting stents (DES) have demonstrated marked efficacy in inhibiting NIH and restenosis, and therefore the need for target lesion revascularisation (TLR) compared to BMS5,6. However, the implications of strut thickness and cell design on DES have still to be determined as their efficacy relies on the ideal combination of key components including: metallic scaffold, drug carrier and antiproliferative agent, which are critical to achieve adequate drug delivery and release at the target site7. Importantly, despite the overall efficacy demonstrated by older-generation DES5,6, persistent concerns regarding deliverability, effectiveness and especially long-term safety8,9 have led to the development of new DES systems including low-profile platforms, drug carriers (durable or non-durable) with optimised biocompatibility, and potent antiproliferative drugs10-15. Our objective was to report the initial results of a novel DES technology incorporating an ultra-thin platform, a bioabsorbable polymer, and a potent pharmacological agent (sirolimus). It has been considered that such stent design could facilitate deliverability and deployment, especially in complex subsets, without compromising safety and efficacy.
Methods
TRIAL DESIGN AND PATIENT POPULATION
The meriT-1 trial was a prospective, non-randomised, single-arm, first-in-man clinical evaluation of the performance of the novel BioMimeTM sirolimus-eluting stent (SES) (Meril Life Sciences Pvt. Ltd., Gujarat, India) in the treatment of non-complex coronary lesions. The objective was to provide preliminary safety, feasibility and efficacy evaluations of this new technology in humans with coronary artery disease treated at a single clinical institution (Lifecare Institute of Medical Sciences and Research, Ahmedabad, Gujarat, India). Key inclusion criteria were: patients >18 years of age; symptoms of stable or unstable angina, or silent ischaemia with a positive functional test for ischaemia; presence of single de novo lesion ≤19 mm in length (by visual estimation) located in native coronary vessel with reference diameter (RD) 2.5 to 3.5 mm (by visual estimation); diameter stenosis (DS) ≥50% and <100% (by visual estimation); TIMI flow ≥2; acceptable candidate for coronary bypass surgery; and agreement to undergo all protocol evaluations including angiographic FU. Key exclusion criteria were: acute myocardial infarction (MI) <72 hours; renal insufficiency with baseline serum creatinine >2.0 mg/dL; previous PCI <30 days to index procedure; left ventricular ejection fraction <30%; left main stem; moderate or severe calcium; severe tortuosity or angulation; thrombus; bifurcation with a side branch ≥2.0 mm; aorto-ostial location; total occlusion; in-stent restenosis; any clinical or non-clinical condition leading to non-compliance with dual antiplatelet therapy (DAPT) during study duration; and known illness or any serious clinical condition with life expectancy <2 years.
The study complied with the Declaration of Helsinki regarding investigation in humans, and was approved by the local ethics committee at the participating institution. All patients provided written informed consent prior to enrolment. The meriT-1 trial was registered at the National Institute of Medical Statistics, Indian Council of Medical Research (Clinical Trials Registry - India [CTRI]) at www.ctri.nic.in/Clinicaltrials: REFCTRI-2009000496, and at the United States National Institute of Health at www.clinicaltrials.gov: NCT01507519.
STUDY DEVICE
The BioMime SES is built on the CE mark-approved Nexgen™ L605 cobalt-chromium stent platform (Meril Life Sciences Pvt. Ltd., Gujarat, India), which combines ultra-thin struts (65 µm) with a unique hybrid cell design comprising a mix of open and closed cells in order to provide optimal radial strength (recoil <3%) without compromising flexibility (Figure 1A). Additional stent specifications include: 6, 8 and 10 crown configurations; Y-connectors and S-links ensuring absence of longitudinal stent compression (or foreshortening); and metal-to-artery ratio ranging from 14% to 16%. The delivery system includes a 0.014” guidewire-compatible semi-compliant rapid exchange balloon catheter made of nylon material with a PTFE- (polytetrafluoroethylene) coated stainless steel hypotube shaft, which includes two radiopaque markers underneath the balloon that are used to delimit its working length. The balloon is designed with abrupt shoulders and minimal overhang (once crimped) to minimise balloon-related edge injury; also, it allows mediated stent expansion from middle (centre) to edges during deployment (Figure 1B).
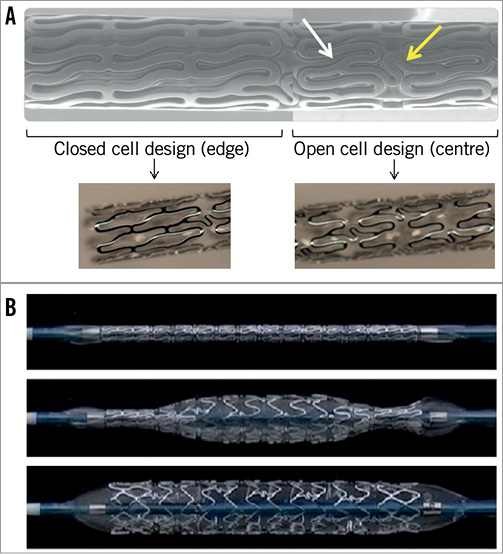
Figure 1. A) Illustration of the BioMime SES design demonstrating its hybrid configuration including closed cell at edges and open cell in the middle (centre). Image on top shows crimped device (SEM image with 50x magnification) with Y-connectors (white arrow) and S-links (yellow arrow). B) Illustration of mediated stent expansion feature: crimped stent (top image); partial expansion starting in the centre portion (image in the middle) followed by full expansion extending to proximal and distal ends or edges (image at bottom). SEM: scanning electron microscope.
The drug-carrier component of the BioMime device consists of a proprietary co-polymer combination (BioPolyTM) of biocompatible and bioabsorbable polymers: PLLA (poly-L-lactic acid) and PLGA (poly-lactic-co-glycolic acid). Overall, this co-polymer formulation provides uniformly thin coating (2 µm) which degrades in approximately 60 days after implantation (data on file at Meril Life Sciences Pvt. Ltd., Gujarat, India). The properties and mechanisms of action of sirolimus, the pharmacological agent used in BioMime, have previously been detailed elsewhere16. In brief, sirolimus exhibits potent immunosuppressive and anti-inflammatory properties, as well as potent suppression of vascular smooth muscle cell proliferation due to cell cycle arrest at the G1-S interface. For the BioMime DES system, sirolimus is coated in a dosage of 1.25 µg per mm2 of stent surface area, given that the total drug dose for a 3.0×19 mm stent would be approximately 121 µg.
PRECLINICAL STUDIES
The pharmacokinetics of the BioMime SES were assessed in a study using a rabbit iliac model performed at the CV Path Institute, Inc., Gaithersburg, MD, USA. In this analysis, 20 stents were implanted in bilateral iliac arteries in 10 animals, and results demonstrated that around 58% of the drug was released in the first three days, with an additional 30% of drug released by 14 days; at 28 days, nearly all the drug was released (data on file at Meril Life Sciences Pvt. Ltd., Gujarat, India) (Figure 2). Furthermore, the histopathologic and histomorphometric evaluations comparing the BioMime SES vs. a control polymer-coated only stent vs. the CypherTM SES (Cordis Corporation, Miami Lakes, FL, USA) in a porcine model were performed using a previously described methodology17,18. Overall, there were 48 stents implanted (BioMime 26; control polymeric 13; Cypher 9) in 27 animals. At 28 days, mean % diameter stenosis (DS) and neointimal thickness were: 19.4% vs. 25.8% vs. 20.6% (p=not significant [NS]), and 0.20 mm vs. 0.31 mm vs. 0.48 mm (p=NS), respectively; at 90 days, results were: 14.2% vs. 26.6% vs. 52.9% (p<0.05), and 0.22 mm vs. 0.42 mm vs. 0.92 mm (p=NS); for BioMime vs. control polymeric vs. Cypher, respectively. Regarding inflammation and injury scores, both BioMime and control polymeric devices showed positive and comparable results including low scores at 28 days (0.22 vs. 0.00, 0.09 vs. 0.23) and at 90 days (0.00 vs. 0.33, 0.17 vs. 0.44), respectively. Also, Cypher showed a higher deposit of fibrinogen at seven days, higher fibrin and necrosis scores at 28 days, and higher inflammation score at 90 days as compared to BioMime and control polymeric, given that such features were virtually absent in both BioMime and control polymeric. Lastly, mean endothelialisation scores with BioMime at days 7, 28 and 90 were 2.94, 3.0 and 2.96, respectively, suggesting near complete strut coverage at early FU (study performed at the Division of Animal Pathology, Division of Cardiology, University of Turin, Turin, Italy; data on file at Meril Life Sciences Pvt. Ltd., Gujarat, India).
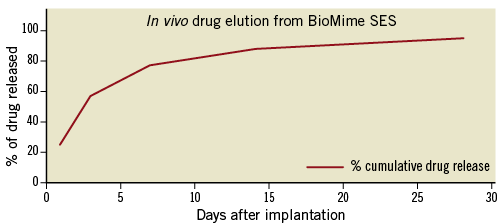
Figure 2. Drug release kinetics of the BioMime SES system.
PROCEDURE
PCI procedure was performed according to current standard guidelines19. Pre-treatment of the target lesion with any devices other than a regular balloon catheter used in angioplasty was not allowed, but direct stenting was allowed. For this study, the BioMime SES device was available in the following range of sizes: 13, 16, 19 and 24 mm in length for diameters of 2.5, 3.0 and 3.5 mm. Multi-stenting and multivessel procedures were not allowed; however, in case of a bail-out situation, a second study stent could be placed either proximally or distally overlapping with the primary stent. A routine 12-lead electrocardiogram was obtained before procedure, immediately post procedure, and 24 hours later. Blood sample laboratory analysis included CK and CK-MB before procedure (<24 hours), <24 hours after procedure, and daily thereafter until hospital discharge.
During procedure, intravenous heparin (70-100 units/Kg) was administered after sheath insertion to maintain an activated clotting time >250 seconds (or >200 seconds if glycoprotein IIb/IIIa inhibitors were used, at the operator’s discretion). Regarding DAPT, aspirin 100-325 mg was given >24 hours prior to procedure if not already taken; for thienopyridine (clopidogrel), a loading dose of 300 mg was given >24 hours prior to procedure (or 600 mg at least two hours prior to procedure) if not already taken (75 mg daily). After the procedure, all patients received aspirin (100-325 mg) daily indefinitely and clopidogrel 75 mg was prescribed daily for at least six months.
ENDPOINTS AND CLINICAL FOLLOW-UP
The primary safety endpoint was the rate of major adverse cardiac events (MACE) at 30 days after procedure. The primary efficacy endpoint was in-stent late lumen loss (LLL) at angiographic FU at eight months. Secondary endpoints were MACE at all study time points up to 24 months and binary restenosis at eight-month angiographic FU. Other predefined endpoints included stent thrombosis (ST) up to 24 months and other quantitative coronary angiography (QCA) parameters at post-procedure and eight-month angiographic FU. MACE was defined as the composite endpoint of cardiac death, MI and ischaemia-driven TLR. Target vessel revascularisation (TVR) included any new revascularisation to the target vessel including TLR. By protocol, all deaths were considered cardiac unless a non-cardiac cause could be clearly established by either clinical assessment or pathological study. MI was classified according to its type (Q-wave or non-Q-wave), and occurrence (periprocedural, spontaneous or post-CABG), following standard definitions as previously reported. ST was classified according to the definitions of the Academic Research Consortium20. Angiographic success was defined as residual stenosis <20% plus final TIMI flow 3 after PCI with the study device. Procedural success was defined as angiographic success plus absence of MACE during index hospitalisation.
Clinical FU was scheduled at 1, 8, 12 and 24 months. All patients were assigned for angiographic FU at eight months. Baseline, procedural and clinical FU data were collected by dedicated clinical research associates; data monitoring was performed physically at the participating clinical site. Data management was performed by an independent clinical research organisation (Integrity Health Services, Mumbai, India). In the current analysis, we report the procedural results, eight-month angiographic FU and 12-month clinical FU.
ANGIOGRAPHIC ANALYSIS
After intracoronary administration of nitroglycerine, serial angiographic studies were obtained in two orthogonal matching views pre- and post-procedure, and at eight-month FU. Angiographic analyses were performed off-line according to standard methodology for DES13 by experienced operators at an independent angiographic core laboratory (Cardiovascular Research Center, São Paulo, Brazil), with a validated 2-D software for QCA analysis (QAngio XA version 7.2; Medis, Leiden, The Netherlands). The minimum lumen diameter (MLD) and the mean reference diameter (RD), obtained from averaging 5 mm segments proximal and distal to the target lesion location, were used to calculate the diameter stenosis (DS=[1–MLD/RD]x100). Acute gain was the change in MLD from baseline to post-stent implantation; late lumen loss (LLL) was the change in MLD from the post-stent implantation angiogram to FU. Overall, QCA measurements were reported as “in-stent” within the stented segment, and “in-segment”, spanning the stented segment plus the 5 mm proximal and distal peri-stent areas.
STATISTICAL ANALYSIS
Categorical data were presented as frequencies (percentages of the total). Data sets with continuous variables were tested for normal distribution with the Shapiro-Wilk normality test. In case of normal distribution, data were presented as mean values±standard deviation (SD); when non-normal distribution was evidenced data were presented as median values with interquartile range [25%, 75%].
Results
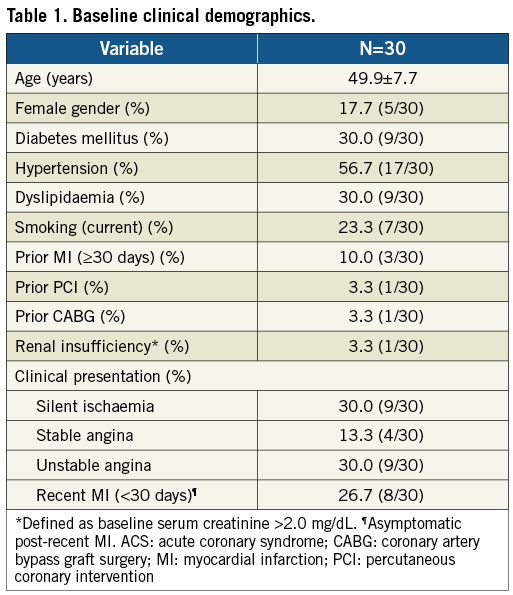
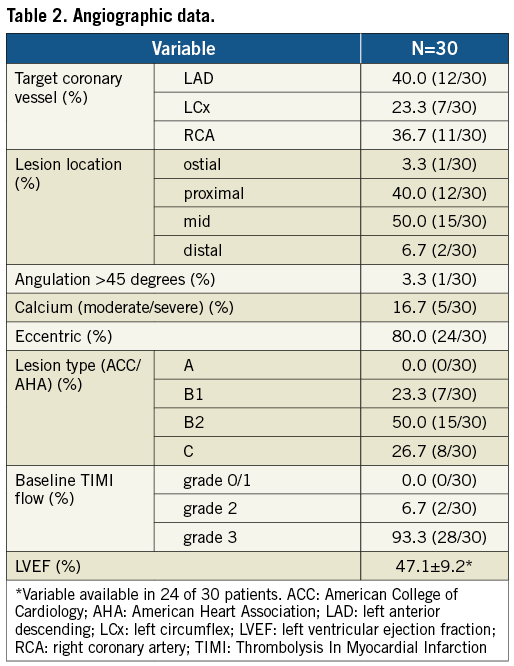
A total of 30 patients were enrolled between March 2009 and February 2010. Mean age was 49.9 years, 30% were diabetics (non-insulin-dependent only), and 36.7% (11/30) had previous MI (eight of 11 asymptomatic post-recent MI <30 days) (Table 1). LAD was the most prevalent target vessel, and the majority of lesions had moderate complexity (Table 2). Procedural outcomes are depicted in Table 3. Radial access was used in approximately 1/3 of cases; the majority of lesions were predilated (90%); there was only one stent implanted per lesion; and angiographic success was 100%. In addition, there were no adverse events during index hospitalisation (procedural success 100%).
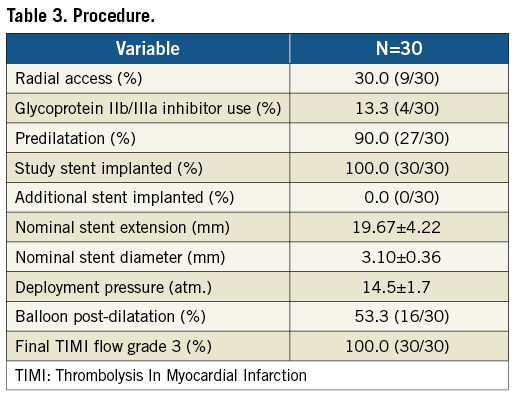
Baseline and final QCA are presented in Table 4. Median lesion length, RD and % DS pre-procedure were 15.51 mm, 2.94 mm, and 80.5%, respectively. Post procedure, median in-stent % DS was <10%. Angiographic FU was available in 86.7% of patients (26/30), since four (asymptomatic) patients refused angiographic re-evaluation. Median in-stent LLL (primary endpoint) was 0.15 mm; also, there were no cases of binary restenosis reported as well as no exaggerated NIH at the edges outside the stent (Table 5). Figure 3 illustrates the cumulative distribution curve for MLD.
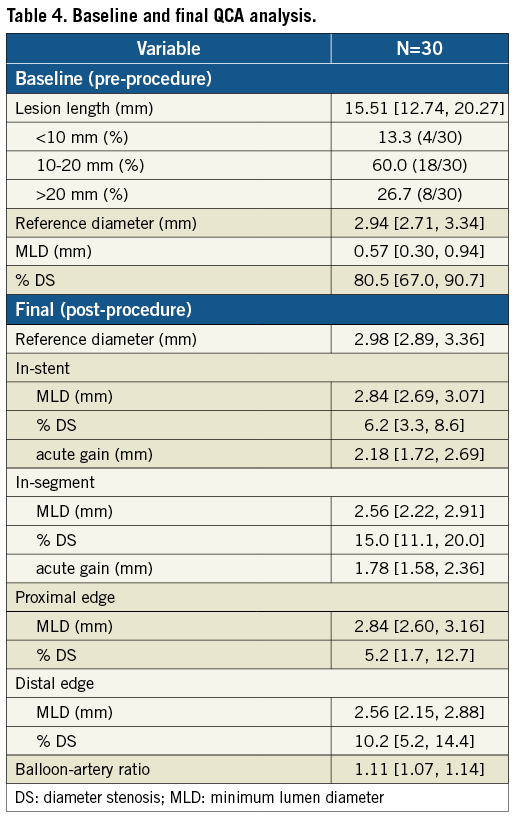
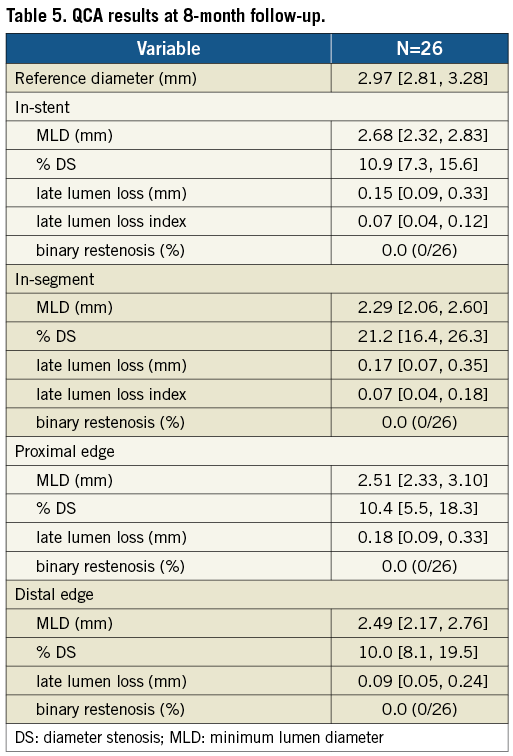
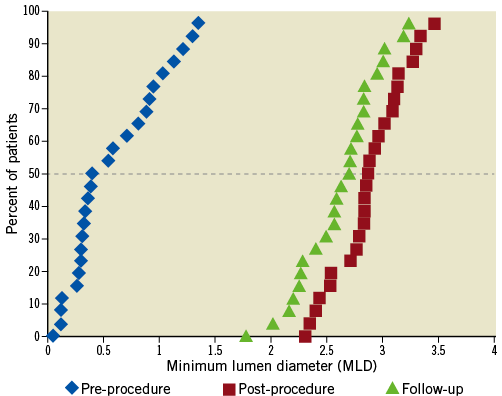
Figure 3. Cumulative frequency distribution curve for MLD at baseline, final (in-stent) and FU (in-stent) for patients with serial angiographic analysis (n=26).
Clinical FU up to 12 months was completed in all patients, and there was neither MACE nor ST (ARC) reported.
Discussion
In the current analysis, the novel BioMime SES with ultra-thin struts demonstrated favourable outcomes in single de novo coronary lesions including: a) high angiographic and procedural success (100%) in lesions with moderate complexity (mainly type B); b) relatively low in-stent LLL (0.15 mm) at mid-term angiographic FU (87%); and c) absence of MACE or ST up to 12 months. Interestingly, a relatively young population (~50 years) with a relatively high prevalence of diabetes (30%) was present, representing the high prevalence of coronary artery disease found in adults in India21. Also, most type C lesions had a baseline lesion length measured marginally over 20 mm by independent QCA analysis, despite visual estimation according to inclusion criteria (≤19 mm), as assessed by operators during the index procedure.
The impact of stent design on outcomes has been already reported. In the ISAR-STEREO trial, 651 patients from daily practice were randomly assigned for PCI with stents with similar design but different strut thickness: thin-strut group (ACS Multi-Link 50 µm) versus thick-strut group (ACS Multi-Link RX Duet 140 µm) (both devices manufactured by Guidant, Santa Clara, CA, USA). At six-month angiographic FU (77%), QCA parameters favoured the thin-strut group (LLL 0.94 vs. 1.17 mm, p=0.001; restenosis 15 vs. 25.8%, p=0.003). Correspondingly, ischaemia-driven TLR at one year was also lower in the thin-strut group (8.6 vs. 13.8%, p=0.03)3. In the subsequent ISAR-STEREO-2 trial, 611 patients were randomly assigned for PCI with stents with different design and strut thickness. In this study, the thin-strut group was treated with ACS RX Multi-Link (interconnected ring design, 50 µm) versus the thick-strut group with Bx VELOCITY® (closed cell design, 140 µm) (Cordis Corporation, Miami, FL, USA). At six-month angiographic FU (78%), there was significantly lower LLL (0.93 vs. 1.19 mm, p<0.001) and restenosis (17.9 vs. 31.4%, p<0.001) with thin struts with open cell design versus thick struts with closed cell design. At one year, TVR due to restenosis was found in 12.3% in the thin-strut group versus 21.9% in the thick-strut group (p=0.002)4. In general, thicker struts appear to induce more NIH with uncoated stents1-4; hence, a similar behaviour would be intuitively anticipated with coated devices. However, such linear correlation has not been clearly demonstrated with DES, as a potent antiproliferative local drug effect may counterbalance or even annul the negative impact of strut thickness on NIH. A randomised study by Pache et al comparing CypherTM SES (Bx VELOCITY platform) versus thin-strut (76 µm) uncoated BeStentTM 2 (Medtronic, Santa Rosa, CA, USA) in 500 patients showed marked reductions in LLL (0.14 vs. 0.94 mm, p<0.001) and restenosis (8.3 vs. 25.5%, p<0.001) with SES versus thin-strut BMS, respectively; also, a significant reduction in TVR was observed with SES (7.2 vs. 18.8%, p<0.001)22. Notwithstanding this, deliverability issues (especially in complex anatomies) as well as stent fracture leading to PCI failure have been seen with older (first) generation DES including Cypher SES23,24. Therefore, most newer-generation DES systems developed to date have incorporated low-profile cobalt-chromium platforms with open cell designs in order to reduce strut thickness, enhance flexibility and deliverability, and improve outcomes10,12,14,15,25. On another topic, the degree of vascular injury caused during PCI procedure may also impact on outcomes. A study by Costa et al (n=1,557) demonstrated that longitudinal and axial injury within the treated segment during stent implant (all Cypher SES) was a common finding (66.5%) and associated with MI (p=0.03) and TVR (p=0.04) up to one year26. The novel BioMime device has one of the smallest strut thicknesses (65 µm) compared to other approved DES systems. In addition, it incorporates a balloon-expandable cobalt-chromium alloy with hybrid design and mediated expansion properties in order to enhance flexibility and avoid vascular injury (Figure 1B). In meriT- 1, median balloon-artery ratio was 1.11 and in-stent LLL at eight months was 0.15 mm with neither exaggerated NIH at the edges nor restenosis reported. Such outcomes are comparable to first-in-man evaluations of the most effective DES systems tested to date11,12,27.
Furthermore, biocompatible polymers appear to be vital constituents of DES since their absence has been related to DES failure due to poor drug delivery and uncontrolled release kinetics28. Durable polymers have been used in first and second-generation DES systems5,6,10,12,14; however, their presence (mainly first-generation DES) has been associated with marked local inflammatory response over time that could lead to intense tissue proliferation or delayed healing and extensive vessel enlargement29. Even though the genesis of ST has been well recognised as multifactorial30, we may speculate that the drug carrier’s everlasting presence may play a role. Moreover, previous studies comparing second-generation DES with biodegradable polymer vs. first-generation DES with durable polymer demonstrated significant reduction in very late ST with DES with biodegradable polymer31. Nonetheless, whether such technology (bioabsorbable polymeric drug carrier) can influence DAPT duration is still unknown and needs further evaluation. For BioMime, a bioabsorbable polymer is used, but the impact of this technology in late outcomes has yet to be determined.
Limitations
In the current analysis, there were no safety concerns up to 12 months, but the relatively low number of patients precludes any definite conclusions regarding safety beyond the perspective of a preliminary single-centre feasibility study. Angiographic results at eight-month FU (87%) suggested high efficacy of the BioMime SES in inhibiting NIH, but serial IVUS analysis was not available.
Conclusions
The novel BioMime SES demonstrated good performance in single coronary lesions including high procedural success and efficacy, as demonstrated by the relatively low late lumen loss (a surrogate of neointimal hyperplasia) at eight-month angiographic FU. Overall, there were no safety concerns in this preliminary evaluation including absence of MACE or ST up to 12 months.
Conflict of interest statement
R. A. Costa has received a research grant from Meril Life Sciences. S. Bhatt is a full-time employee of Meril Life Sciences. The other authors have no conflicts of interest to declare.
