Abstract
Background: Polymer-free drug-coated stents aim to avoid the inflammatory potential of durable polymers, thereby improving the long-term safety profile, and allowing a shorter duration of dual antiplatelet therapy.
Aims: The BIOVITESSE study was conducted to assess the safety and clinical performance of the BIOrapid polymer-free coronary stent system coated with a novel highly lipophilic sirolimus derivate.
Methods: BIOVITESSE was a prospective, multicentre, first-in-man study that enrolled subjects with de novo coronary lesions in two cohorts of 33 patients each. The primary endpoint of the first cohort was strut coverage at one month as assessed by optical coherence tomography. The primary endpoint of the second cohort was late lumen loss at nine-month follow-up.
Results: Patients were on average 63 years old (range: 42-87) and 12% had diabetes. The 66 patients had 70 lesions with an average lesion length of 12.5±5.4 mm. Predilatation was performed in 91.4% and post-dilatation in 87.1% lesions; device success was obtained in 97.4%. At one month, 95.2±5.6% (95% CI: 93.2-97.2) of struts were covered and at nine months, in-stent late lumen loss was 0.31±0.30 mm (95% CI: 0.20-0.42) and in-segment late lumen loss was 0.20±0.29 mm. Two target lesion failures occurred (3.1%): one at day 1 (to cover an asymptomatic stent edge dissection), and one at day 288 post-procedure for restenosis. No stent thrombosis was reported during the 12-month study duration.
Conclusions: The BIOrapid stent system exhibited an excellent safety profile, high strut coverage at one-month, and moderate angiographic efficacy according to the late lumen loss at nine-month angiographic follow-up.
Introduction
Durable polymer drug-eluting stent (DES) coatings have shown suboptimal biocompatibility and negative effects on vessel healing due to chronic inflammation and local toxicity, potentially leading to proliferative and thrombogenic responses over time. Furthermore, the safety of DES appears to be dependent on relatively long (≥6 months) dual antiplatelet therapy (DAPT), carrying the risk of bleeding123.
To overcome these limitations and to allow a shorter DAPT duration after percutaneous coronary intervention (PCI), polymer-free drug-coated stents (DCS) were introduced to mitigate the disadvantages of a permanent polymer. DCS combine the advantages of drug delivery with the long-term safety profile of a bare metal stent (BMS)1245, because once the drug is delivered to the surrounding tissue only the BMS remains in the vessel. Several different types of DCS have been tested, most of them characterised by a modification of the stent surface through nanopores, micropores or macropores, or microholes which act as drug reservoirs567.
The BIOrapid drug-coated coronary stent system (Biotronik AG, Bülach, Switzerland) is a novel polymer-free spray-coated ultrathin stent with a novel sirolimus derivate that has a higher lipophilicity than sirolimus or other sirolimus derivates used in currently available DES and, as a result, the drug has a higher tissue penetration. The stent backbone is not altered and is the same that is used for the PRO-Kinetic Energy BMS and the Orsiro DES (both Biotronik; strut thickness of 60 µm for stents ≤3.0 mm and 80 µm for >3.0 mm)8. The latter has proven to be safe and effective, and non-inferior89 or even superior1011 to current DES standards.
Angiographic and histological data from animal studies indicate lower late lumen loss (LLL) and diameter stenosis than the ProKinetic Energy BMS platform, and similar LLL and diameter stenosis to the Orsiro DES, with similar inflammation scores (data on file). The BIOrapid Vascular ImplanT Abluminal PolymerlESs Safety and Efficacy (BIOVITESSE) study intended to assess the safety and performance of this novel device in humans, with early stent strut coverage assessed at one month by optical coherence tomography (OCT), and angiographic late lumen loss at nine months as a surrogate endpoint for efficacy.
Methods
STUDY DESIGN
BIOVITESSE was a prospective, multicentre, first-in-man trial including patients with symptomatic ischaemic heart disease due to discrete de novo lesions.
The full list of inclusion and exclusion criteria is provided at the www.ClinicalTrials.gov website (NCT03263858). Main inclusion criteria were age ≥18 and ≤85 years, stable or unstable angina pectoris, documented silent ischaemia or haemodynamically stable non-ST-elevation myocardial infarction, a maximum of 2 single discrete de novo lesions in 2 separate native coronary arteries, reference vessel diameter of 3.0-3.8 mm and a target lesion length of up to 22 mm. Main exclusion criteria were left main disease, chronic total occlusion, bifurcation lesions requiring side branch intervention (if side branches >2 mm were involved), or prior treatment with a drug-coated balloon. Sixty-six patients were enrolled in 5 centres in Switzerland (33 in cohort 1 and 33 in cohort 2). After reaching the primary endpoint for cohort 1, an interim report was submitted to SwissMedic and cohort 2 commenced after SwissMedic approval. Clinical follow-up took place at 1, 9, and 12 months.
The study was conducted according to applicable local regulations, the Declaration of Helsinki, ISO14155:2011, and was approved by the sites’ ethics committees and competent authorities. All patients provided informed consent. Monitoring included 100% source document verification, an independent angiographic and OCT core laboratory (MedStar Health Research Institute, Washington, D.C., USA) assessed all imaging parameters, an independent clinical events committee adjudicated all adverse events, and an independent data safety monitoring board reviewed all serious adverse events, device and procedure failures and any device-related adverse events. It further permitted the start of the second BIOVITESSE cohort after review of the 30-day outcomes from the first cohort.
STUDY DEVICE AND PROCEDURE
BIOrapid has a thin drug coating of 2-3 µm that consists of BIOTORCIN™ (Biotronik), a highly lipophilic mTOR inhibitor, a derivative of sirolimus, blended with an inert non-polymer excipient. The drug is released over a period of approximately three months (Supplementary Figure 1). Stent sizes available for the study were as follows: diameter 3.0 and 3.5 mm, and length 15, 20, and 26 mm. The rapid-exchange delivery system comes with a polyamide 12 semi-compliant balloon which is the same as used for the PRO-Kinetik Energy BMS and the Orsiro DES (both Biotronik).
Predilatation using a balloon diameter 0.5 mm smaller than the reference vessel diameter and a balloon length equal to or shorter than the target lesion was required. Post-dilatation was optional. Dual antiplatelet therapy was recommended according to the current European Society of Cardiology (ESC) guidelines3 and standard of care at the study centres.
ENDPOINTS AND DEFINITIONS
The primary endpoint of cohort 1 was strut coverage at one month post-procedure assessed by OCT, the primary endpoint of cohort 2 was in-stent LLL at nine months assessed by quantitative coronary angiography (QCA). Secondary endpoints were device success, defined as final residual diameter stenosis <30% by QCA, using the assigned device only and successful delivery of the stent, appropriate stent deployment, and successful removal of the device. Clinical endpoints were target lesion failure (TLF), a composite of cardiac death, target vessel myocardial infarction, and clinically driven target lesion revascularisation (TLR), target vessel failure (TVF), a composite of cardiac death, target vessel myocardial infarction and target vessel revascularisation (TVR), all cause death, myocardial infarction (periprocedural being adjudicated according to the Society of Cardiovascular Angiography and Interventions [SCAI] definition and spontaneous according to the universal and extended historical definitions)121314, and stent thrombosis15. Angiographic endpoints were in-segment LLL, diameter stenosis and binary restenosis, and OCT endpoints included stent strut data at 1 month (cohort 1) and 9 months (cohort 2) (Supplementary Appendix 1).
STATISTICAL ANALYSIS
This analysis is based on the intention-to-treat population based on the data available. Clinical endpoints are calculated using Kaplan-Meier statistics. Descriptive statistics include the mean and standard deviation and median and interquartile ranges as applicable. For categorical (qualitative) variables, the absolute and relative frequencies are displayed; 95% confidence intervals (CIs) were calculated as applicable. Statistical calculations were performed using SAS version 9.4 (SAS Institute Inc., Cary, NC, USA).
Results
From September 2017 to March 2018 (cohort 1) and from June 2018 to January 2019 (cohort 2), 66 patients with 70 lesions were enrolled (Figure 1). Patients were 63±11 years on average (range: 42-87 years), 12.1% had diabetes mellitus, and 34.8% previous myocardial infarction (Table 1). Per core laboratory assessment, lesions were 12.5±5.4 mm long and more than half were type B2/C lesions (65.2%) (Table 2).
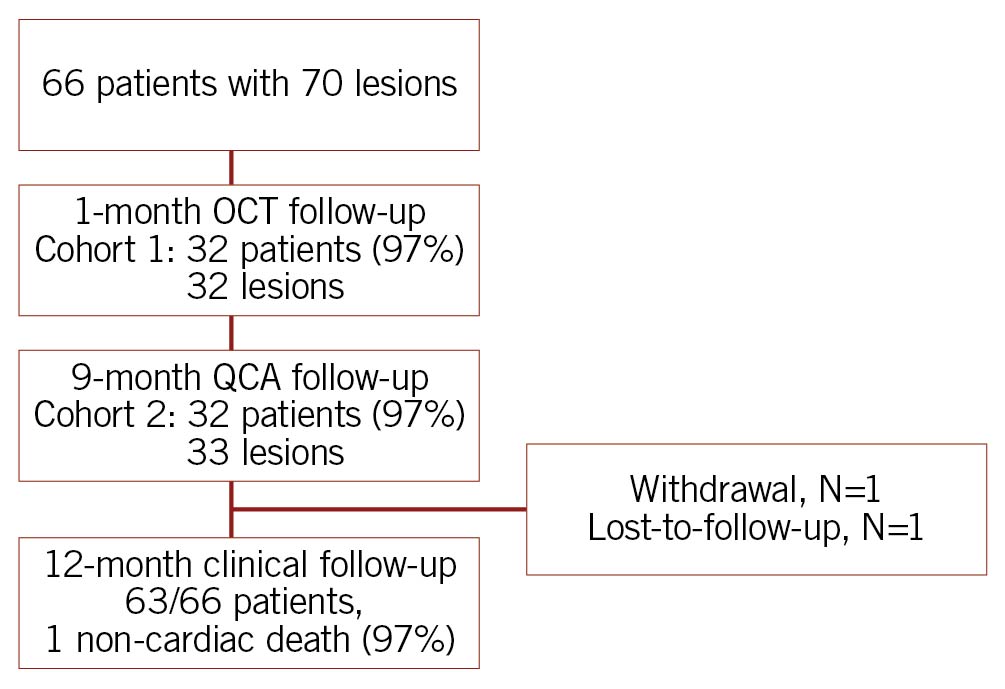
Figure 1. Study flow chart. One patient excluded due to suboptimal image quality. OCT: optical coherence tomography; QCA: quantitative coronary angiography
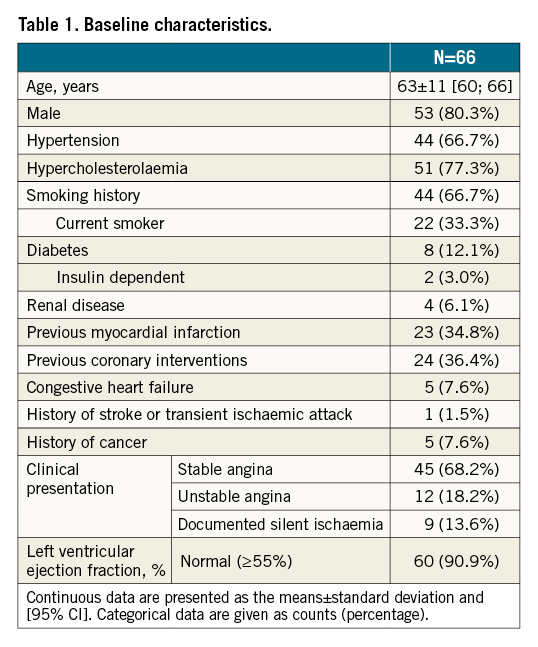
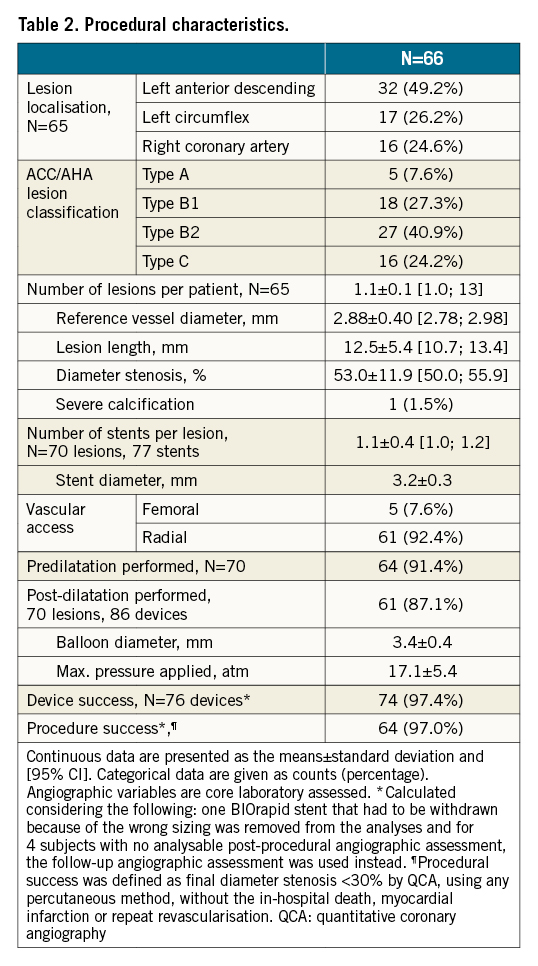
Nearly all lesions were predilated (91.4%), and post-dilatation was performed in 87.1%. One stent was not deployed because the operator decided to change to another stent size. Device success was obtained in 97.4% (Table 2).
OCT assessment was successfully obtained in 97% of patients of cohort 1 at one month. Stent strut coverage amounted to 95.2±5.6% (95% CI: 93.2-97.2); 1.4±3.0% of struts were uncovered and malapposed. At 9 months, strut coverage of cohort 2 amounted to 98.9±2.1% (95% CI: 98.1-99.6) (Table 3). Angiographic analyses at 9 months were available for 97% of patients of cohort 2 and showed an in-stent late lumen loss of 0.30±0.31 mm (95% CI: 0.20-0.42), median 0.24 (IQR: 0.10-0.44). Approximately 20% of LLL values were above 0.5 mm (Table 4, Central illustration).
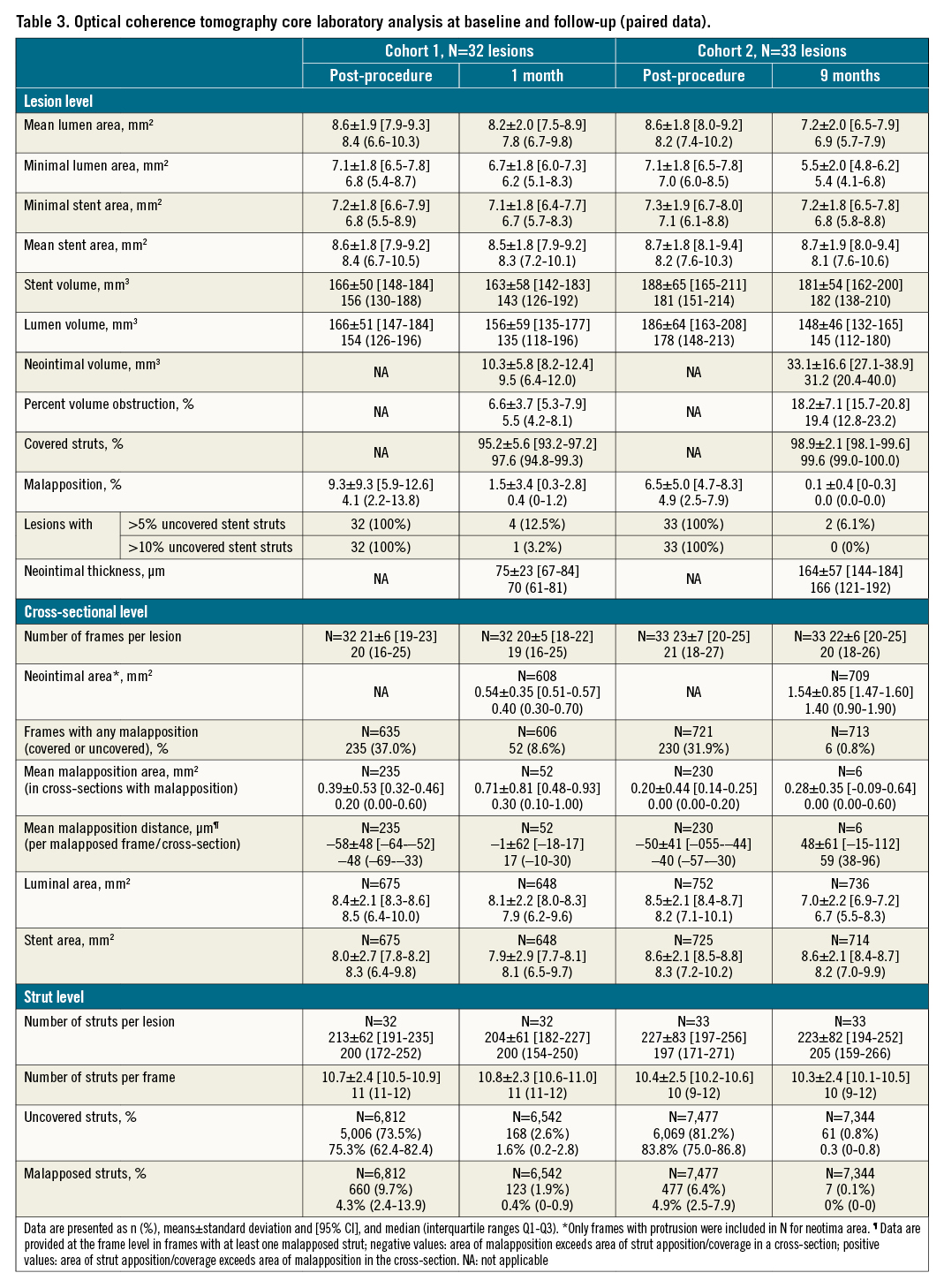
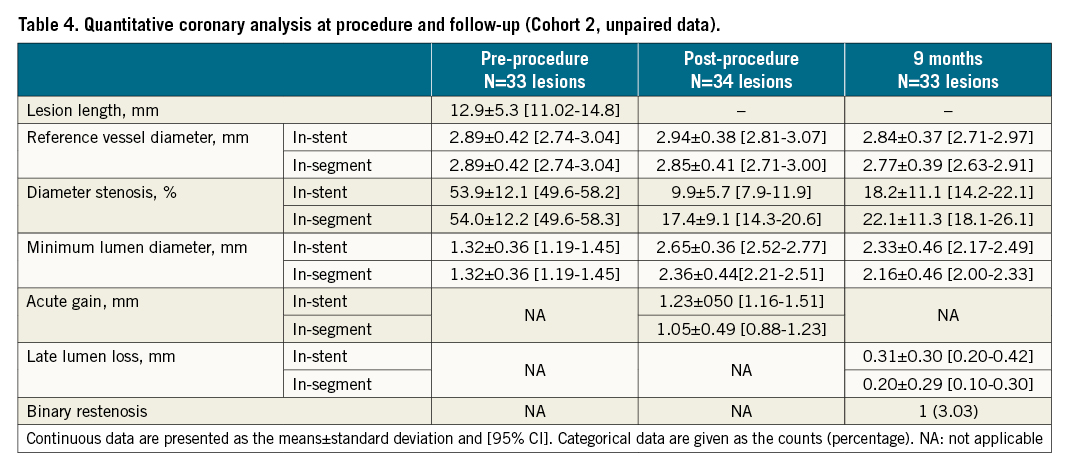
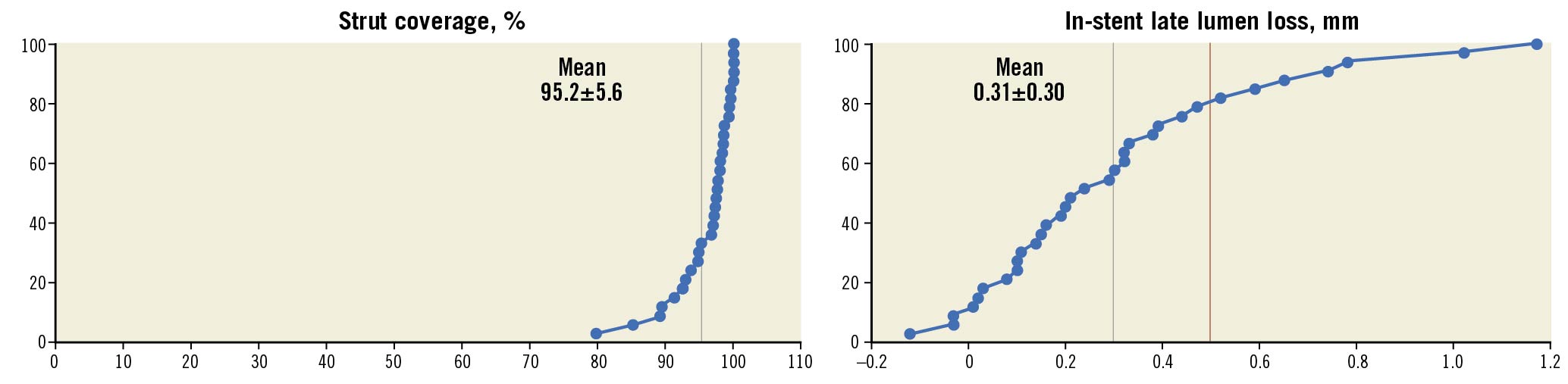
Central illustration. Primary endpoint strut coverage at one month (cohort 1) and in-stent late lumen loss at nine months (cohort 2) by core laboratory assessment. The black line reflects the mean LLL and the red line the 0.5 mm threshold above which the LLL is considered clinically relevant29. Strut coverage is based on patient level. LLL: late lumen loss
Cardiovascular medication is presented in Supplementary Table 1. At 1 month, 90.8% (59/65) of patients were on DAPT, at 9 months 67.2% (43/64) and at 12 months 41.3% (26/63).
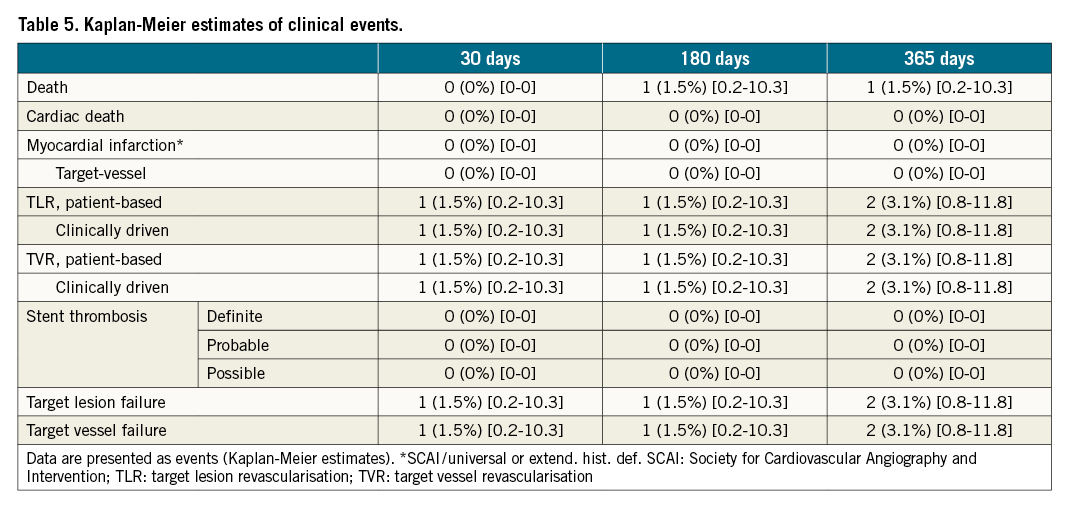
Two TLF occurred during the 12-month study duration (Table 5). One proximal edge dissection during the index procedure treated the following day was adjudicated as TLR by the clinical events committee. The second TLR occurred on post-procedure day 288 in a patient with recurrent angina and a 70% restenosis detected during elective nine-month follow-up angiography. No cardiac death, myocardial infarction, or definite, probable or possible stent thrombosis was observed. There was one non-cardiac death on day 58 due to progredient respiratory failure in the context of a competing pulmonary disease.
Discussion
The use of the novel BIOrapid drug-coated coronary stent system in the BIOVITESSE study resulted in a nearly complete strut coverage (95.2%) at an early time point of 1 month, moderate angiographic efficacy at 9 months, and low clinical event rates with no thrombotic events.
The study was conducted thoroughly, with 100% monitoring, clinical events committee adjudication, core laboratory assessment, and high follow-up compliance.
STRUT COVERAGE
Nearly all struts were covered at 1 month (95.2%), comparing well to the stent strut coverage at 1 month of the PRO-Kinetic BMS (93%), which forms the backbone of the BIOrapid stent16. In contrast, the Orsiro DES, which uses the same stent backbone, but with a sirolimus-eluting biodegradable polymer cover, exhibits a strut coverage of 80.4% at 1 month17.
Our study further demonstrated good strut coverage when compared to contemporary stents. At 1-month post-procedure, strut coverage was 88% for the Resolute Integrity™ (Medtronic, Minneapolis, MN, USA)18, 85% for Ultimaster™ (Terumo Corp., Tokyo, Japan)19, and 88% for Resolute Onyx™ (Medtronic)20. Similar outcomes (91.4% strut coverage) have been reported for the polymer-free drug-filled stent (DFS)21. This nearly complete strut coverage may allow to safely stop DAPT early in patients with high bleeding risk or simple lesions with low thrombotic risk. However, complete coverage, as determined by OCT, does not always mean vessel healing, because OCT cannot differentiate between fibrin and true endothelial coverage. Furthermore, the current results were obtained in simple lesions and may not be directly applicable to more complex lesion subsets. A comparison of baseline parameters is provided in Supplementary Table 2.
ANGIOGRAPHIC EFFICACY
An improvement in in-stent LLL (0.31±0.30 mm, median 0.24) compared with the PRO-Kinetic BMS backbone (0.88±0.58 mm at 6 months in the MULTIBENE study)22 could be observed, showing a drug effect. However, LLL was higher than for the Orsiro DES (0.10±0.32 mm and 0.05±0.02 mm 9-month LLL in the BIOLFLOW-II and -VI studies)2324 and other contemporary DES such as the XIENCE DES (Abbott) (0.11±0.29 mm and 0.07±0.02 mm LLL in the BIOLFLOW-II and -IV studies), similar to the Ultimaster DES (0.26±0.35 mm)232425, and higher than postulated in the systemic review from the ESC-European Association of Percutaneous Cardiovascular Interventions (EAPCI) task force (LLL of 0.18 mm [IQR: 0.13-0.25] for new DES and 0.16 mm [IQR 0.13-0.22] for new FDA-approved DES)26.
The LLL results were, however, similar to other polymer-free DES. The BioFreedom US trial reported an LLL of 0.32±0.53 mm (median 0.19)7, the BioFreedom first-in-man trial reported LLL medians of 0.17 (IQR 0.12-0.35) and 0.19 (IQR 0.07-0.58) for the high- and low-dose formulation27. For the Medtronic polymer-free DFS, an LLL of 0.26±0.28 mm was reported21, and for the COBRA Polyzene-F NanoCoated stent (NCS; CeloNova BioSciences, Inc., TX, USA) an LLL of 0.84±0.48 mm28.
Whether the current high stent strut coverage and moderate late lumen loss represents a sweet spot between high degree of early safety and sufficient angiographic potency remains to be demonstrated in larger clinical trials. A recent patient-level meta-analysis considers LLL values of ≤0.5 as clinically negligible and suggests the cut-off value of 0.5 mm as an objective performance criterion2629.
Whether the applied drug concentration of novel sirolimus-derivate on the BlOrapid stent system should be increased can be speculated, e.g., the nominal active drug dose on the BlOrapid stent was around eight times lower than on the BioFreedom stent27 and approximately three times lower than on the Orsiro stent (data on file) for similar stent sizes. Likewise, the BioFreedom stent showed a large variance in 9-month LLL in the low-dose formulation with an interquartile range of 0.07 to 0.58 mm and inferior 12-month TLR rate in the low-dose application (6.7% versus 1.7%), leading to a discontinuation of the low-dose DCS in that study. Notably, this effect weakened over time, with a 5-year TLR rate of 10.8% in the low-dose application versus 13.4%27.
It can also be assumed that the total dispersion of the drug through the stent over a period of approximately 3 months was not sufficient to prevent further hyperplasia between 3 months and 9 months after the procedure.
CLINICAL OUTCOMES
Clinical outcomes were promising with the absence of ischaemic thrombotic events such as stent thrombosis or target vessel myocardial infarction. Further, only one restenosis-associated TLR occurred. However, this is a first-in-man trial with relatively simple lesions and TLR rates could increase in more complex lesions considering the moderate LLL observed.
Limitations
This first-in-man trial has several limitations. The device has been studied in a selected patient population with relatively simple coronary lesions. Moreover, around two thirds of patients were still on DAPT at 9-month follow-up. The study is not randomised and can only be compared against historical controls. The small patient number prevents a thorough analysis of the root cause(s) for the higher than expected LLL. Considering the fact that this is the first assessment of a novel sirolimus derivate administered in a low dose, in hindsight, a higher dose should have been tested simultaneously, as done for the BioFreedom DCS27.
Conclusions
In this first-in-man trial evaluating the novel polymer-free abluminally drug-coated BIOrapid stent system, it was shown that, up to 12 months, the system is safe and clinically effective in treating de novo coronary artery lesions, with only one restenosis-associated TLR (1.5%) and no thrombotic events. Near complete stent strut coverage was achieved at one month with 95.2% of struts covered. Late lumen loss confirms the antirestenotic efficacy of the novel sirolimus derivate as compared to LLL for the same platform without the drug. However, results suggest a lower efficacy for this iteration than observed with modern thin-strut DES.
Impact on daily practice
This is the first trial of the polymer-free BIOrapid stent system and the first human exposure to a low-dose highly lipophilic sirolimus derivate coated on a vascular implant. The BIOVITESSE study demonstrates that the device is safe and effective, but with regard to late lumen loss it was not competitive with contemporary drug-eluting stents, particularly considering that impaired outcomes may be expected in more complex patients and lesions. Given the promising clinical outcomes and high strut coverage at one month, it might be worthwhile to assess a device iteration with a higher drug dose.
Acknowledgements
The authors thank Beatrix Doerr for her help in preparing the manuscript, reimbursed by Biotronik.
Funding
The study was sponsored by BIOTRONIK CRC, Inc., La Jolla, CA, USA. The sponsor was involved in the study design, conduct of the study, and statistical analysis. Furthermore, the sponsor reimbursed the medical writer who worked under the guidance of the coordinating author. The sponsor was not involved in the decision to submit the manuscript.
Conflict of interest statement
L. Räber has received research grants to the institution by Abbott, Biotronik, Boston Scientific, Heartflow, Medis, Sanofi, and Regeneron, and speaker/consultation fees from Abbott, AstraZeneca, Amgen, Canon, Sanofi, Occlutech, and Vifor. J. Häner has received a travel grant from Bayer. T. Luescher has received research and educational grants from Abbott, Ablative Solutions, Amgen, AstraZeneca, Boehringer Ingelheim, Novartis, Sanofi, Servier, and Vifor, and honoraria from Ablative Solutions, Amgen, Daiichi Sankyo, Novo Nordisk and Sanofi. M. Moccetti has received educational and research grants from Biotronik, Abbott, Boston Scientific, Microport, and Biosensors. M. Roffi has received institutional research grants from Medtronic, Biotronik, Boston Scientific, Terumo and GE Healthcare. S. Stortecky has received research grants to the institution from Edwards Lifesciences, Medtronic, Abbott Vascular and Boston Scientific, speaker fees from Boston Scientific, and consultant fees from BTG/Boston Scientific and Teleflex outside the submitted work. H. Garcia-Garcia declares that his institution receives research grants from Biotronik, Medtronic, Boston Scientific, Abbott, Neovasc, Corflow, Shockwave, Chiesi, and Philips. R. Waksman is on the Advisory Board of Abbott Vascular, Amgen, Boston Scientific, Cardioset, Cardiovascular Systems Inc., Medtronic, Philips, and Pi-Cardia Ltd., is a consultant of Abbott Vascular, Amgen, Biotronik, Boston Scientific, Cardioset, Cardiovascular Systems Inc., Medtronic, Philips, Pi-Cardia Ltd., and Transmural Systems, receives grant support from AstraZeneca, Biotronik, Boston Scientific, and Chiesi, is on the speakers bureau of AstraZeneca and Chiesi, and is an investor in MedAlliance and Transmural Systems. P. Siegrist reports personal consultant fees/honoraria/speaker’s bureau from Boston Scientific, SMT, Shockwave Medical, Philips, Abiomed, Astra Zeneca, and Amgen, institutional grant support/research contract from Biotronik and Terumo, and personal stock options of Merck & Co. The other author has no conflicts of interest to declare.
Supplementary data
To read the full content of this article, please download the PDF.
