Abstract
Aims: To report the six-month angiographic and two-year clinical outcome data from the first-in-man study with the Ultimaster DES, a thin-strut cobalt-chromium sirolimus-eluting stent (SES) with an innovative abluminal-gradient-coated bioresorbable polymer.
Methods and results: CENTURY is a multicentre, single-arm, prospective study that enrolled 105 patients (113 lesions) with coronary artery disease. All patients were scheduled to have an angiographic follow-up at six months, while 45 and 20 patients respectively had IVUS and OCT assessments. The primary endpoint was six-month in-stent late lumen loss. Secondary endpoints included clinical, IVUS and OCT outcomes. Clinical follow-up is available up to two years and will continue up to five years. Procedural success was 97.1% and device success was 100%. Angiographic late loss at six months was 0.04±0.35 mm, also reflected in a low binary restenosis rate of 0.9% and confirmed by IVUS-assessed neointimal volume obstruction of 1.02±1.62%. The mean strut coverage assessed by OCT was 96.2% with 1.66±4.02 malapposed stent struts. There were no deaths in the study, three (2.9%) periprocedural and one (0.9%) spontaneous myocardial infarction, not related to the target vessel. At one and two years, the target lesion failure rate was 3.8% and 5.7%, while the TLR rate was 1.9% and 2.8%, respectively. There was one acute definite stent thrombosis.
Conclusions: The Ultimaster™ novel bioresorbable polymer sirolimus-eluting stent demonstrated good performance, including high procedural success and strong suppression of neointimal proliferation at six months. Good safety and effectiveness were shown up to two years in the studied population.
Introduction
Drug-eluting stents (DES) have demonstrated an undisputed superior antirestenotic efficacy, associated with improved clinical outcomes, over bare metal stents (BMS)1-3. However, their early positive results were challenged by less favourable long-term safety data4,5. The reported higher risk of late and very late stent thrombosis was attributed to the intrinsic characteristics of first-generation DES, such as the platform design and/or the polymer. This has led to a marked improvement in stent design and an increasing interest towards the development of novel drug-carrier systems including bioresorbable polymers and non-polymeric stent surfaces6,7. The bioresorbable polymers, in particular, are expected to reduce inflammatory triggers for the vessel wall and as such reduce late thrombosis and restenosis8,9. In addition, shortening polymer degradation time might allow an earlier and safer withdrawal of dual antiplatelet therapy (DAPT).
In this study, we report on a new DES, the Ultimaster™ (Terumo Corp., Tokyo, Japan). The Ultimaster is a thin-strut, cobalt-chromium, sirolimus-eluting stent with bioresorbable polymer coated only abluminally by applying special gradient technology. The aim of the CENTURY study was to assess the safety and performance of the Ultimaster stent in patients with coronary artery disease.
Methods
PATIENT POPULATION
Patients who were at least 18 years of age with ischaemic heart disease due to de novo lesions in up to two native coronary arteries were considered for enrolment. Major clinical exclusion criteria were: myocardial infarction (MI) within the preceding 72 hours; intolerance to aspirin, heparin, clopidogrel, ticlopidine, sirolimus or similar drugs, contrast media, cobalt-chromium or nickel and any bleeding or coagulation disturbances (a complete list of inclusion and exclusion criteria can be found in the Online Appendix). The main angiographic exclusion criteria were total occlusion, left main or ostial target lesion, severe calcification, evidence of thrombus or severe tortuosity.
The study patients were enrolled in eight medical centres (Online Appendix) from November 2011 to February 2012. The study was conducted according to the Declaration of Helsinki, Good Clinical Practice, ISO 14155, and respecting all country-specific regulatory requirements. The protocol was reviewed and approved by the ethics committee of each participating medical institution. Prior to any test or procedure related to the trial, the benefits and the risks of the study were explained, and written informed consent, approved by the ethics committee, was obtained from each participating patient.
THE ULTIMASTER CORONARY STENT SYSTEM
The Ultimaster coronary stent system consists of a cobalt-chromium (Co-Cr) bare metal stent platform featuring thin struts (80 µm) with a unique open-cell design for easy access to a side branch and conformability to the vessel wall. The stent is mounted on a rapid-exchange catheter with a high-pressure, semi-compliant balloon. The Ultimaster platform is coated with sirolimus (3.9 µg/mm stent length) in a matrix with bioresorbable, Poly (DL-lactide-co-caprolactone) polymer. A thin biocompatible, bioresorbable gradient coating was the key to the innovative design of the system (Figure 1). Gradient coating is intended to reduce polymer cracking and delamination on the hinges of the stent. The drug coating components were chosen to optimise performance with minimal drug and polymer content and controlled drug release kinetics. Within three to four months the polymer is either metabolised, through DL-lactide and caprolactone into carbon dioxide and water or excreted in urine and faeces. A reduced drug dose was possible thanks to an abluminal (outside surface) coating which ensures an equal amount of drug delivered to the target tissue, as is the case with double the drug dose on DES with a circumferential coating. Furthermore, coating only the abluminal surface leaves the luminal side of the stent free from drug and polymer, as such enhancing endothelial coverage.

Figure 1. Image of gradient coating of Ultimaster (magnified 200 times).
The drug release profile is adjusted to best match the biological response: initial release will suppress injury and inflammation induced by the catheter manipulation and stent implantation. The remaining drug will be released simultaneously with polymer bioabsorption within three to four months. Thereafter, the Ultimaster DES is expected to become a bare metal stent. The Ultimaster DES is available in diameters of 2.5, 2.75, 3.0, 3.5 and 4.0 mm, and in length sizes of 12, 15, 18, 24 and 28 mm.
CORONARY STENTING PROCEDURE
It was recommended that all patients receive a medication regimen according to the routine practice of the hospital. After mandatory predilation, an appropriately sized stent was implanted. Additional study stents were permitted for edge dissection or other suboptimal results. Post-dilation was at the operators’ discretion. Pre-procedural and post-procedural ECGs were obtained and cardiac enzymes were measured at baseline, and 12 to 24 hours after the procedure or at discharge, whichever came first. Dual antiplatelet therapy was mandatory for six months after stent implantation.
Patient follow-up
All surviving patients were to have a repeat angiography at six months±15 days, along with intravascular ultrasound (IVUS) on a pre-specified subset of 40% of patients and optical coherence tomography (OCT) in 20% of patients (Figure 2). The selection of the centres for IVUS and OCT was based on availability of the corresponding technology. Clinical follow-up was scheduled at 30 days, six and 12 months and yearly thereafter up to five years. The clinical follow-up included assessment of angina status, monitoring of cardioactive and antithrombotic drug use, interim hospitalisations, occurrence of major adverse cardiac events, and any invasive and non-invasive diagnostic test or interventional treatment that had occurred since the previous contact.
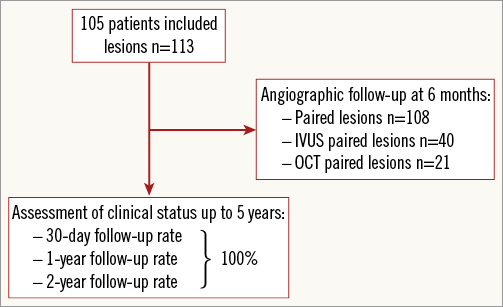
Figure 2. CENTURY study flow chart.
Study management
A data monitoring committee (DMC) was responsible for the review of all data and identification of potential safety issues. An independent clinical events committee (CEC) reviewed and adjudicated all major adverse cardiac events. The members of the committees were not affiliated to the study sponsor and were not participating in the trial. The study was managed by an independent contract research organisation (Genae, Antwerp, Belgium) responsible for monitoring, data management and analysis. Data were collected and stored on an independent electronic data collection platform (Merge Healthcare, Chicago, IL, USA) with 100% on-site source data verification.
Quantitative coronary angiography, IVUS and OCT evaluation
An independent core laboratory (Cardialysis, Rotterdam, The Netherlands) analysed all angiographic, IVUS and OCT recordings using validated methodologies.
Endpoints and definitions
The primary endpoint was angiographic in-stent late loss at six months post procedure defined as the difference between the post-procedure minimal lumen diameter (MLD) and the follow-up angiography MLD. Secondary clinical endpoints were assessed at one, six, 12 and 24 months and will continue yearly up to five years and include: target lesion failure (TLF), defined as the composite of cardiac death, target vessel-related myocardial infarction (MI) and clinically indicated target lesion revascularisation (TLR); stent thrombosis (ST) according to Academic Research Consortium (ARC) definitions10. All secondary angiographic, IVUS and OCT endpoints were assessed at six months and included: angiographic in-stent and in-segment binary restenosis rate; in-stent, in-segment, proximal, and distal MLD; IVUS-assessed neointimal hyperplasia volume and % volume obstruction; OCT-assessed stent strut coverage and malapposition. The majority of the endpoints were defined according to the Academic Research Consortium (ARC) and can be found in the Online Appendix10.
Sample size calculation and statistical analysis
The primary analysis of this study takes into account that this is a single-arm trial, using the primary endpoint of late loss at six months. The main hypothesis of the study was that Ultimaster would show superiority in in-stent late loss at six months versus its historical control, the bare metal stent platform Kaname® (Terumo Corp). The assumption for the study was late loss for the Kaname stent of 0.90±0.50 mm and 0.30±0.50 mm for the Ultimaster (the data for Kaname were not available at the time of study design). The late loss reduction by at least 0.50 mm from the one observed in the Kaname stent was considered a clinically significant improvement. To prove the superior performance of the Ultimaster DES vs. the Kaname stent, a one-sided 97.5% confidence interval of the six-month in-stent late loss was constructed. In order to observe this difference with a power of 80%, a maximum extent of 0.20 mm of mean confidence interval was taken into account.
The primary endpoint and all secondary endpoints were analysed on the intention-to-treat population. Clinical events including death, MI and revascularisation are reported on a per patient basis. For patients with multiple lesions, a failure of any lesion was counted towards the composite event rate. For the angiographic endpoints, all treated lesions were analysed. For continuous variables, differences between the treatment groups were examined by analysis of variance, while Fisher’s exact test was used for categorical variables. All statistical analyses were performed using SAS statistical software, version 8 (SAS Institute Inc., Cary, NC, USA).
Results
CLINICAL CHARACTERISTICS
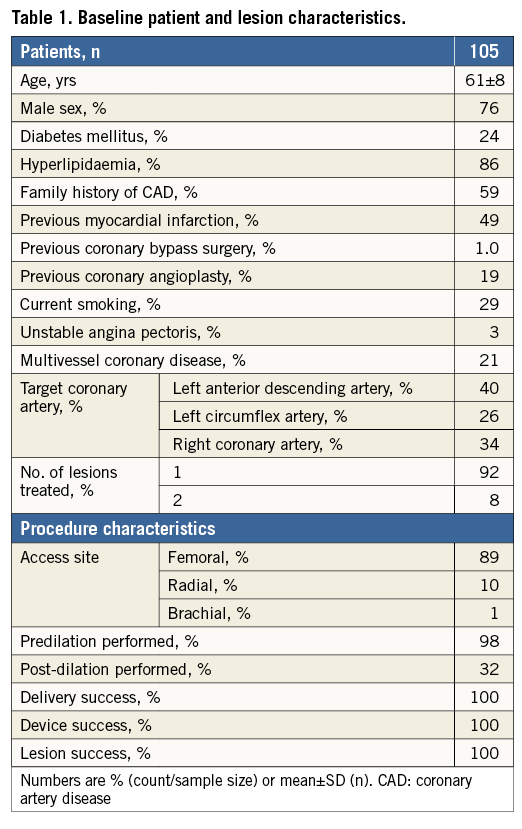
In the CENTURY study, 105 patients were enrolled and 113 lesions were treated by the Ultimaster stent. Baseline demographics are presented in Table 1. The mean age of the study population was 61±8 years, 76% were males and 24% of patients had diabetes mellitus. Prevalent cardiac risk factors were rather high for a FIM study, with 49% of patients with previous MI and 21% of patients with multivessel disease. Delivery, device and lesion success were all 100%, while procedure success was 97%.
QUANTITATIVE CORONARY ANGIOGRAPHY FINDINGS
Angiographic follow-up at six months was performed in 101 (96%) patients with 108 (96%) Ultimaster-treated lesions (Table 2). The Ultimaster stent proved to be superior in six-month late lumen loss to its bare metal platform historical control, the Kaname stent, with 0.04±0.35 vs. 0.75±0.43 mm (p<0.001 for superiority) (Figure 3). The binary in-stent and in-segment restenosis rates with the Ultimaster stent were low at 0.9% and 1.9%, respectively.
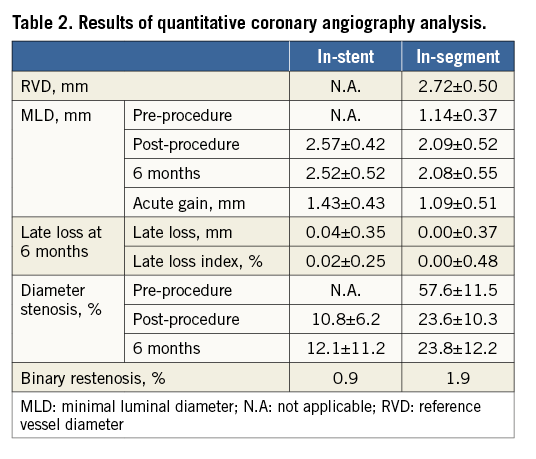

Figure 3. Primary endpoint: in-stent late loss at 6 months.
INTRAVASCULAR ULTRASOUND AND OPTICAL COHERENCE TOMOGRAPHY EVALUATION
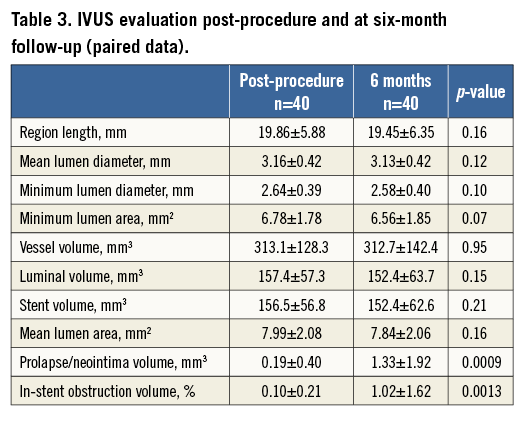
Intravascular ultrasound was used to assess vessel, stent, and lumen volume, plaque volume, and in-stent volume obstruction after Ultimaster stent implantation in 45 (39%) lesions at baseline and in 40 (35%) lesions at six-month follow-up, respectively. IVUS-calculated parameters reflect angiographic findings with a mean plaque volume and plaque area of 1.3±1.9 mm3 and 0.04±0.15 mm2, respectively, at six months (Table 3).
OCT analysis was performed in a total of 20 patients with 21 (18.5%) lesions analysed (Table 4). The measured lumen volume and stent volume at six-month follow-up were similar to IVUS findings taking into account that only half of the IVUS patients also had OCT. An incomplete stent strut apposition (ISA) of 1.66±4.02% was found with an average ISA volume of 1.86±6.58 mm³ per lesion. The mean strut coverage at six months was 96.2% (median 98%). The strut coverage distribution is shown in Figure 4.
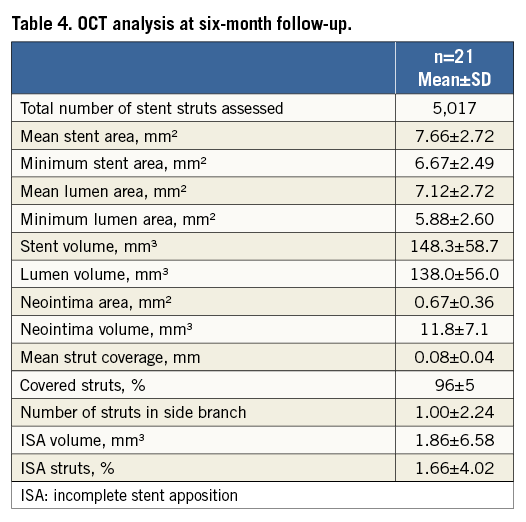
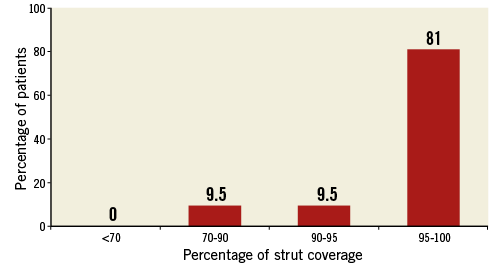
Figure 4. Stent strut coverage distribution.
Clinical outcomes
IN-HOSPITAL OUTCOMES
During the index hospitalisation two (1.9%) patients suffered periprocedural non-Q-wave MIs. One patient with untreated dissection developed stent thrombosis shortly after stent implantation and underwent target lesion revascularisation with stent implantation.
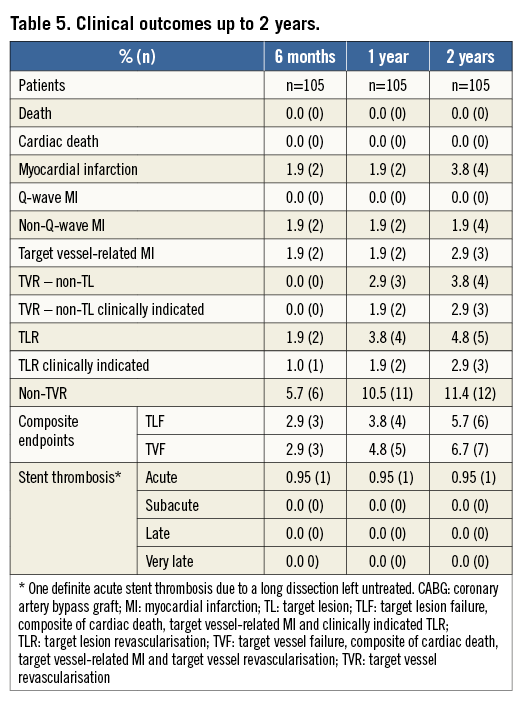
SIX-MONTH FOLLOW-UP
Clinical follow-up at six months was completed for all patients. Between hospital discharge and six months (195 days) there were no deaths or new MIs (Table 5). During angiographic follow-up one patient underwent target lesion revascularisation. The clinical events committee adjudicated this event as non-clinically indicated TLR, as the diameter stenosis was 37% and the patient was asymptomatic. The rate of TLF at six months was 2.9%, and 97% of patients were on DAPT.
ONE- AND TWO-YEAR FOLLOW-UP
All patients attended one- and two-year follow-up. Between six months and one year there were no deaths, MIs, or ST (Table 5). There were two TLRs (1.9%), one being clinically indicated (0.9%), three TVR non-TLRs (2.9%), two being clinically indicated (1.9%). The cumulative TLF rate at 12 months in Ultimaster-treated patients was 3.8%. There were no new stent thromboses reported. Between one year and two years, two patients experienced new events. One patient had revascularisation of the target lesion. The same patient experienced periprocedural increase of cardiac enzymes adjudicated as myocardial infarction. One patient had spontaneous increase of cardiac enzymes 13 months after stent implantation. Control angiography showed a fully patent study stent, without any signs of narrowing.
At one and two years, 66% and 9% of patients were still on DAPT and 92% and 93% of patients were free from angina, respectively (data not shown).
Discussion
In this prospective, first-in-man study the Ultimaster DES showed very low late loss at angiographic follow-up of six months and good two-year clinical performance. The main findings of the CENTURY study are: 1) Ultimaster DES is superior to its bare metal platform Kaname stent with respect to in-stent late loss, resulting in a 95% late loss reduction; 2) Ultimaster DES showed low in-stent and in-segment restenosis; 3) IVUS and OCT data support the favourable efficacy profile of Ultimaster, revealing a very thin and homogenous layer of neointima covering the stent struts six months after stent implantation, reaching an average strut coverage rate of 96% (median: 98%); 4) the clinical benefit of Ultimaster was reflected in low rates of clinically indicated revascularisations of target lesions up to two years.
The primary endpoint of the CENTURY study was in-stent late loss at six months post stent implantation. The main rationale to select a surrogate rather than a clinical endpoint in this study was that, in the months following stent implantation, LL as an angiographic parameter is proven to be a reliable predictor of the long-term clinical efficacy of DES and a strong, monotonically related predictor of binary restenosis and TLR11. As the low rates of TLR accomplished by most DES would mandate large patient populations to prove the clinical non-inferiority or superiority of a new device in direct comparisons, the use of angiographic surrogates to draw clinical conclusions, based on the findings of Pocock et al, appears clinically sound and objective11. The in-stent LL of 0.04 mm at six months closely resembles the 0.04 mm in the pivotal CYPHER DES trials, but also the LL reported for contemporary DES using limus drugs such as XIENCE® 0.12 mm (Abbott Vascular, Santa Clara, CA, USA), Biomatrix™ 0.26 mm (Biosensors International Pte Ltd, Singapore), Nobori™ 0.10 mm (Terumo Corp., Tokyo, Japan) (at nine months), Orsiro 0.05 mm (Biotronik AG, Bülach, Switzerland) (at eight months), SYNERGY 0.10 mm (Boston Scientific, Natick, MA, USA) or PROMUS Element™ 0.15 mm (Boston Scientific)8,12-16. This good efficacy of Ultimaster DES can be explained by the Ultimaster abluminal coating, which directs most of the drug towards the target tissue rather than being washed out into peripheral circulation from the luminal surface as in the case of DES with circumferential coating.
Delayed healing, hypersensitivity reaction to polymer carriers, and insufficient restoration or non-functional endothelium, along with specific patient/lesion characteristics, have been implicated in the well-known drawbacks of first-generation DES, such as stent thrombosis, restenotic late catch-up and vessel remodelling17-19. The majority of these negative effects can be, to some extent, attributed to, or are dependent on, polymer properties. As Mehilli et al20 reported in their study, biodegradable polymers showed superior antirestenotic efficacy compared to durable and polymer-free stents, without the long-term negative effects of persistent coverings. The OCT and IVUS investigations of the Ultimaster stent were consistent with these assumptions. The IVUS analysis confirmed the absence of vessel remodelling with unchanged vessel volume peri-stent (313.10±128.30 and 312.67±142.44 mm3 post stent implantation and at six-month follow-up, respectively). Unlike prior studies showing positive remodelling with some DES21, in the CENTURY study there were no signs of vessel enlargement, ectasia or aneurysms observed at follow-up. This suggested that there were no reactions of the vessel to the bioresorbable polymer or to the sirolimus drug after implantation of the Ultimaster stent. Other IVUS parameters corresponded to data obtained with newer-generation limus-eluting stents. In-stent neointimal volume obstruction for the Ultimaster of 1.02±1.62% compares favourably with data reported in the SPIRIT II trial for XIENCE (2.77±5.02%)22 or in the EVOLVE trial for the PROMUS Element (3.40±5.06%) and SYNERGY stents (2.68±4.60%)16.
OCT analysis of the Ultimaster stent showed a low rate of incomplete stent apposition. These results are similar to the findings reported by Guagliumi et al for the XIENCE and PROMUS Element stents at six months (1.66% in Ultimaster, 1.80% for XIENCE and 1.51% for PROMUS Element)23. Strut coverage at six months in the Ultimaster DES was above 96%, while it was 91.5% and 94.1% in the PROMUS Element and XIENCE DES, respectively23. In the RESOLUTE All Comers study which randomised the Resolute zotarolimus-eluting stent versus the XIENCE everolimus-eluting stent, at 13 months strut coverage was 92.6% and 94.2%, respectively24. The patients enrolled in the latter study had higher lesion complexity, possibly explaining the differences.
Stent thrombosis (ST) has become one of the most critical issues in the daily use of DES, particularly in patients non-compliant to DAPT4. Shorter polymer bioresorption time might offer more safety in case of abrupt disruption or shorter duration of DAPT without increasing the risk of ST9. Although not powered to examine this clinically important phenomenon, the CENTURY study showed favourable results with no late or very late stent thrombosis up to two-year follow-up, despite only a fraction of patients being on DAPT. As polymer bioresorption is expected to be complete by three to four months, the Ultimaster stent is expected to maintain its good safety profile in the longer term, this hypothesis deserving further investigation in a dedicated trial.
Shorter polymer degradation time has been mentioned as a cause for concern due to a potential inflammatory response to polymer degradation products. This issue has been addressed in numerous preclinical studies in rabbits and pigs implanted with the Ultimaster stent for 14 and 28 days, three, six and nine months. There were no indications of increased inflammatory response at any studied time point (data on file at Terumo). The data from Ultimaster-implanted vessels were compared either with a bare metal stent platform or with the XIENCE stent, adding further evidence to the safety of the bioresorbable polymer coated on the Ultimaster stent. As polymer bioresorption is expected to be complete by three to four months, the Ultimaster stent is expected to maintain its good safety profile in the longer term, this hypothesis deserving further investigation in a dedicated trial.
The clinical outcomes of Ultimaster in the CENTURY study are similar to those of other contemporary drug-eluting stents. The rate of TLR of 1.9% at one year closely resembles the results of the platinum-chromium, cobalt-chromium everolimus-eluting stent in a selected population studied in the PLATINUM clinical trial25. The 3.8% TLF rate at one year compares well to the 10.0% TLF rate reported for the Orsiro DES in the BIOFLOW 1 trial15 and 2.7% for the XIENCE DES in SPIRIT II26, both studying similar patient populations. The low rate of adverse events between one and two years is additionally reassuring. The TLF rate at two years (5.7%) in the CENTURY study is similar to those reported for XIENCE in SPIRIT II (6.6%)22 and for PROMUS Element (6.1%) and SYNERGY DES (5.5%) in the EVOLVE trial16.
Study limitations
Despite being rigorously conducted by applying the highest standards for data quality assurance, this study has several important limitations. It was non-randomised and used a historical control for the primary endpoint. Furthermore, the trial was powered for the angiographic endpoint and, as such, due to a small number of patients, does not allow drawing any firm conclusions concerning clinical outcomes. It is important to note that the results from this trial are specific to the relatively low-risk patient population studied and cannot be uncritically generalised to the much broader population of patients with more complex lesions, such as bifurcations, total occlusions, or bypass graft stenosis. Several ongoing trials, enrolling larger numbers and high-risk patients with clinical endpoints, are expected to answer these questions.
Conclusions
In this first-in-man trial, the novel Ultimaster bioresorbable polymer sirolimus-eluting stent demonstrated good performance with low late loss that was translated into low rates of binary restenosis and target lesion revascularisation up to two years, with no late or very late stent thrombosis. The Ultimaster showed good stent strut coverage and good stent apposition at six-month follow-up.
| Impact on daily practice The ease of implantation, potent antiproliferative properties and excellent safety profile make the Ultimaster drug-eluting stent (DES) a valid alternative for treatment of patients similar to those enrolled in this trial. In addition, due to the rapid and complete polymer bioresorption, this DES becomes equivalent to a bare metal stent within three to four months. These features could make the Ultimaster DES an attractive option in patients at high bleeding risk, if ongoing clinical trials demonstrate the safety of early withdrawal of dual antiplatelet therapy. |
Funding
The CENTURY study was funded by the Terumo Corporation, Tokyo, Japan.
Conflict of interest statement
The authors have no conflicts of interest to declare.
Appendix. CENTURY eligibility criteria, study organisation and endpoints and definitions
ELIGIBILITY CRITERIA
INCLUSION CRITERIA
Patient is ≥18 years old.
Patient is eligible for percutaneous coronary intervention (PCI) and an acceptable candidate for coronary artery bypass grafting (CABG).
Clinical evidence of ischaemic heart disease and/or a positive functional study, stable angina pectoris (Canadian Cardiovascular Society [CCS] classification 1, 2, 3 or 4), or unstable angina pectoris (Braunwald Class IB-C, IIB-C, or IIIB-C), or silent ischaemia.
The target lesion(s) or target vessel(s) meet(s) all the following criteria:
a) The target lesion is a single de novo lesion or restenotic post-PTCA (non-stented) lesion in a native coronary artery.
b) The stenosis of target lesion(s) is ≥50% and <100% (by visual estimation).
c) The target lesion length must be ≤25 mm.
d) The target reference vessel diameter must be (by visual estimation) suitable for treatment with stents between 2.5 and 4.0 mm.
Patient has been informed of the nature of the study, understands the study requirements, agrees to its provisions and has provided written informed consent as approved by the Institutional Review Board/Ethics Committee of the respective clinical site.
The patient is willing and able to comply with all specified follow-up evaluations.
EXCLUSION CRITERIA
Most recent left ventricular ejection fraction (LVEF) of the patient is <25%.
Known allergies to the following: aspirin, Clopidogrel bisulphate (Plavix®), Prasugrel (Effient®) or Ticlopidine (Ticlid®), heparin, Sirolimus, cobalt, chromium, nickel, or contrast agent (that cannot be adequately premedicated).
Platelet count is less than 100,000 cells/mm3 or more than 700,000 cells/mm3.
WBC count is less than 3,500 cells/mm3.
Evidence of an ST-segment elevation acute myocardial infarction (MI) or non-ST-segment elevation MI with positive Troponin within 72 hours before the intended treatment.
Previous PCI (within 30 days).
Presence of any other significant lesion of >50% stenosis (by visual estimation) anywhere within the target vessel.
Significant lesions in any non-target vessel that will require interventional treatment within 30 days post procedure.
Planned future interventional procedure in the target vessel.
The target lesion(s) require(s) treatment with a device other than PTCA balloon prior to stent placement (e.g., but not limited to directional coronary atherectomy, excimer laser, rotational atherectomy, thrombus aspiration, etc.).
Previous stenting anywhere within the target vessel(s).
Target vessel has evidence of thrombus.
Excessive tortuosity (>60°) of the target vessel proximal to the target lesion (by visual estimation).
The target lesion(s) has any of the following characteristics (by visual estimation):
a) Ostial or bifurcation lesion (within 3 mm from region of origin of target vessel, by visual estimation).
b) Target lesion involves a side branch >2 mm in diameter.
c) Target lesion has excessive tortuosity (>45°).
d) Moderate to severely calcified lesion which cannot be successfully predilated.
e) Target lesion is located in or supplied by an arterial or venous bypass graft.
f) Significant (>40% by visual estimation) stenosis proximal or distal to the target lesion.
g) A complete occlusion (TIMI flow 0 or 1).
Target lesion is located in the left main trunk.
Stroke or transient ischaemic attack within 180 days prior to the baseline procedure.
Active peptic ulcer or upper GI bleeding within 180 days prior to the baseline procedure.
The patient has bleeding haemorrhagic diathesis or coagulopathy.
The patient will refuse a blood transfusion.
The patient has a widespread peripheral vascular disease.
Acute or chronic renal dysfunction (creatinine >2.0 mg/dl).
The patient requires multiple stent implantations for a tandem lesion.
Life expectancy less than one year.
Patient is currently participating in an investigational drug or device study that has not completed the primary endpoint or that clinically interferes with the current study endpoints.
Note: Trials requiring extended follow-up for products that were investigational, but have become commercially available since then, are not considered investigational trials.
In the Investigator’s opinion patient has (a) comorbid condition(s) that could limit the patient’s ability to participate in the study, compliance with follow-up requirements or impact the scientific integrity of the study.
Patient is in cardiogenic shock.
Female of child-bearing potential.
CENTURY study organisation
CENTURY coordinating investigator: W. Wijns, OLV Ziekenhuis, Aalst, Belgium
The CENTURY Data Monitoring Committee: R. Hoffmann, Vivantes Netzwerk für Gesundheit GmbH, Berlin, Germany; B. Rensing, St. Antonius Ziekenhuis, Nieuwegein, The Netherlands; C. Hanet, CHU Dinant Godinne, Namur, Belgium.
The CENTURY Steering Committee: W. Wijns, OLV Ziekenhuis, Aalst, Belgium; E. Barbato, OLV Ziekenhuis, Aalst, Belgium; P. M. Seferovic, Clinical Centre of Serbia, Belgrade, Serbia; A. Neskovic, Clinical Hospital Centre Zemun, Belgrade, Serbia.
Clinical Event Commitee: R. Hoffmann, Vivantes Netzwerk für Gesundheit GmbH, Berlin, Germany; B. Rensing, St. Antonius Ziekenhuis, Nieuwegein, The Netherlands; C. Hanet, CHU Dinant Godinne, Namur, Belgium.
CENTURY sites and investigators: OLV Ziekenhuis, Aalst, Belgium: E. Barbato, J. Bartunek, B. de Bruyne, G. Heyndrick, M. Vanderheyden, C. Van Mieghem, E. Wyffels; ZOL Sint-Jan, Genk, Belgium: M. Vrolix, P. Selleslagh, J. Dens, J. Van Lierde; ZNA Middelheim, Antwerp, Belgium: S. Verheye, E. Sanidas, G. Van Langenhove, P. Vermeersch, F. Van den Branden, B. Van Reet, C. Convens, P. Van den Heuvel; Clinical Centre Nis, Nis, Serbia: S. Salinger-Martinovic, M. Zivkovic, T. Kostic, M. Damjanovic, S. Apostolovic, M. Pavlovic, N. Krstic, E. Dimitrijevic, N. Bozinovic, Z. Perisic; Clinical Centre Kragujevac, Kragujevac, Serbia: N. Jagic, V. Miloradovic, D. Nikolic, M. Sreckovic; Clinical Centre of Serbia, Belgrade, Serbia: B. Beleslin, D. Orlic, S. Stojkovic, G. Stankovic, V. Vukcevic, V. Dedovic, Z. Mehmedbegovic, M. Zivkovic, B. Terzic, M. Dobric; Clinical Hospital Centre Zemun, Zemun, Serbia: A. Neskovic, I. Ilic, M. Cerovic, S. Kafedzic, A. Vlahovic-Stipac, Z. Stajic, B. Putnikovic, I. Nikolajevic, A. Aleksic, B. Ilisic; Institute for Cardiovascular Diseases Dedinje, Belgrade, Serbia: D. Sagic, B. Petrovic, M. Colic, B. Milosavljevic, R. Babic, L. Mangovski, D. Topic, Z. Tinjic, Z. Antonic.
Endpoints and definitions
Procedural endpoints included lesion success defined as the attainment of <30% residual stenosis by visual assessment and/or <50% by QCA using any percutaneous method; device success defined as achievement of a final diameter stenosis of <50% by QCA, and/or <30% by visual assessment, using the assigned device only; procedure success defined as achievement of a final diameter stenosis of <30% by visual assessment and/or <50% by QCA, using any percutaneous method, without the occurrence of death, MI, or repeat revascularisation of the target lesion during the hospital stay. Any death was considered cardiac, unless clear non-cardiac causes could be determined. MI was defined either as the development of pathological Q-waves in at least two contiguous leads with or without elevated cardiac enzymes or, in the absence of pathological Q-waves, as an elevation in creatinine kinase levels to greater than twice the upper limit of normal in the presence of an elevated level of CK-MB fraction or troponin. Target lesion revascularisation was defined as repeat percutaneous intervention of the stented lesion including 5 mm proximal and distal from the edge of the stent, or bypass surgery of the target vessel that was performed for a clinical indication and was due to restenosis or closure of the target lesion. A revascularisation was considered clinically indicated if prompted by a positive functional study, or ischaemic ECG changes at rest in a distribution consistent with the target vessel, or ischaemic symptoms with an in-lesion diameter stenosis ≥50% by QCA or if lesion diameter stenosis was more than 70% at follow-up, even in the absence of clinical symptoms.
