Abstract
Aims: To report the four-month and nine-month angiographic results as well as one-year clinical follow-up from the first-in-man study with the silicon carbide and sirolimus-eluting bioabsorbable polymer (poly-L-lactic acid (PLLA) polymer) -coated cobalt-chromium Orsiro stent.
Methods and results: A group of 30 patients with documented myocardial ischaemia related to a single de novo coronary stenosis up to 22 mm in length, in vessels with a 2.5 to 3.5 mm reference diameter, and between >50% and <90% diameter stenosis were enrolled at two sites. The primary endpoint of the study was in-stent late lumen loss at nine months. The secondary endpoints included major adverse cardiac events (MACE) at one year defined as the composite of cardiac death, ischaemia-driven target lesion revascularisation (TLR) and target vessel myocardial infarction (MI). Procedural success was 100%. Angiographic late lumen loss was 0.12±0.19 mm and 0.05±0.22 mm at four and nine months respectively. At one-year clinical follow-up, the composite MACE was 10% with one patient who died from cardiac death and two patients who had ischaemia-driven target lesion revascularisation. There was no report of MI or stent thrombosis.
Conclusions: The Orsiro drug-eluting stent demonstrated potency with low rates of in-stent neointimal hyperplasia and cardiovascular events but warrants further evaluation in a larger population cohort with longer follow-up time points.
Introduction
The emergence of drug-eluting stents (DES) has contributed to the reduction of restenosis and the need for repeat revascularisation1-3. However, the first-generation DES were associated with an increased risk of late events including stent thrombosis and late restenosis4-7. Efforts to mitigate these risks have included prolongation of dual antiplatelet therapy, and improvements in stent platforms, polymer carriers and drug selection4,8. The use of a biodegradable polymer has the potential to reduce the sustained inflammatory response of the arterial wall, facilitating re-endothelialisation and minimising the risk of thrombus formation and late restenosis5,6. The Orsiro stent is a hybrid combination of passive and active coatings with the aim of optimising clinical effectiveness and mitigating the risk of late events. The active component facilitates this via a highly biocompatible polymer matrix that delivers sirolimus within the initial 12-14 weeks followed by gentle degradation within 24 months, leaving only the passive silicon carbide coating. The passive coating encapsulates the stent, minimising the interaction between the metal surface and the surrounding tissue.
The aim of the BIOFLOW-I study was to evaluate the safety and efficacy of the Orsiro hybrid drug-eluting stent in patients with single de novo coronary lesions.
Methods
STUDY DESIGN AND PATIENT SELECTION
BIOFLOW-I was conducted at two centres in Bucharest, Romania, with a total of 30 patients being enrolled. The study was conducted in accordance with the Declaration of Helsinki and was approved by the competent authority and site local ethics committees. All patients gave written informed consent before entering the study. All 30 patients were to be followed up with angiography at four and nine months with a subgroup of 15 patients also having an intravascular ultrasound (IVUS) at four and nine months. Clinical follow-up was intended at one month and then at 12, 24 and 36 months. Patients were eligible if they had clinical evidence of unstable or stable angina or documented silent ischaemia. Single de novo lesions in one coronary artery with a reference vessel diameter between 2.5 and 3.5 mm, lesion length ≤22 mm and diameter stenosis between 50% and 90% were allowed to be treated during the index procedure. Exclusion criteria were a left ventricular ejection fraction of ≤30%, three-vessel coronary artery disease, ostial lesion, thrombus in target vessel, significant stenosis in target vessel that might require revascularisation or impede run-off, previous DES or CABG in the target vessel, or any type of PCI to target vessel or side branch in the previous 12 months.
STUDY ENDPOINTS AND DEFINITIONS
The primary endpoint of the study was in-stent late lumen loss at nine months by quantitative coronary angiography evaluated by an independent core lab (Medstar Health Research Institute, Washington, DC, USA). Secondary endpoints were target lesion revascularisation (TLR), clinically driven TLR, and target vessel revascularisation (TVR) at one, four, nine, 12, 24 and 36 months post procedure. Secondary endpoints also included a composite of cardiac death, target vessel myocardial infarction, and clinically driven target lesion revascularisation, and a composite of all-cause mortality, any MI and any revascularisation. All serious adverse events, neointimal hyperplasia volume, in-stent late lumen loss, in-segment late lumen loss, stent thrombosis, binary in-stent restenosis and binary in-segment restenosis, along with device success, procedural success and lesion treatment success were other endpoints.
Device success was defined as a final residual diameter stenosis of <30% by QCA, using the assigned device only. Procedure success was defined as achievement of a final diameter stenosis of <30% by QCA, using any percutaneous method, without the occurrence of death, Q-wave, WHO-defined non-Q-wave, or repeat revascularisation of the target lesion during the hospital stay. Measurement within the stented segment was defined as in-stent, and measurement within 5 mm proximal or distal of the stent edges was defined as in-segment. Late lumen loss was defined as the difference between the minimum luminal diameter post procedure and at follow-up as determined by quantitative angiography.
Target lesion revascularisation was considered clinically-driven if the patient had a positive functional study, ischaemic ECG changes at rest in a distribution consistent with the target vessel, or ischaemic symptoms and an in-lesion diameter stenosis ≥50% by QCA. Revascularisation of a target lesion with an in-lesion diameter stenosis ≥70% (by QCA) in the absence of the above-mentioned ischaemic signs or symptoms was also considered clinically-driven. Major adverse cardiac events (MACE) were defined as the composite of cardiac death, target vessel MI, or clinically-driven target lesion revascularisation (TLR). Recurrent myocardial infarction was defined as any myocardial infarction that occurred after the index procedure9. Stent thrombosis was defined according to the Academic Research Consortium definitions10.
All major adverse cardiac events were adjudicated by an independent clinical events committee (CEC, listed in the appendix section). One hundred percent of source data verification was performed during monitoring.
THE ORSIRO STENT
The PRO-Kinetic Energy stent is a tubular, balloon-expandable stent sculpted by laser from a single tube of L-605 cobalt-chromium (CoCr) alloy. The stent consists of circular segments at each end followed by a transition zone and helicoidally arranged struts in the middle. Each loop of the helix is connected to the next loop by three longitudinal struts. The stent surface is fully coated with a layer of amorphous hydrogen-rich silicon carbide (aSiC:H) acting as a diffusion barrier (PROBIO®), sealing the bare metal surface and reducing ion release. The PROBIO® silicon carbide coating consists of silicon carbide, a ceramic material, a chemical compound of the element silicium (also called silicon) and the element carbon. In clinical trials, this passive coating on a cobalt-chromium stent has shown promising results relative to success rates, angiographic and clinical outcomes11. The polymer compound used as a carrier material for the supply and release of sirolimus is a high molecular poly-L-lactic acid (PLLA). The stent body surface is completely coated by a matrix consisting of the carrier poly-L-lactide (PLLA) and the active substance sirolimus. The matrix coating has an abluminal thickness of 7.5 µm and a luminal coating of 3.5 µm. The drug load is 1.4 µg per mm2 stent surface. The thickness of the coated stent struts is 71 µm for stents with a nominal diameter of ≤3.0 mm and 91 µm for the two larger sizes.
STUDY PROCEDURE
Balloon predilatation of the target lesion was mandatory prior to stent implantation. Success was defined as achievement of a final diameter stenosis of <30% measured by offline QCA using any percutaneous method, without the occurrence of death, MI, or repeat revascularisation of the target lesion during the hospital stay. If post-dilatation was required, a balloon at least 2 mm shorter than the stented segment was used. Post intervention, the patients were recommended to take at least 75 mg aspirin daily and clopidogrel 75 mg (ticlopidine alternatively) for at least 12 months.
FOLLOW-UP
Planned follow-up investigations were hospital visits at one, four, nine, 12, 24 and 36 months. At the four- and nine-month visits an angiographic assessment was scheduled. Clinical status assessment included the ischaemic/anginal status according to the Canadian Cardiovascular Society Classification of stable angina, any (serious) adverse events, current concomitant medication including antiplatelet/anticoagulant therapy, a physical examination, a functional test (e.g., a bicycle stress test or ECHO), and a 12-lead ECG.
QUANTITATIVE CORONARY ANGIOGRAPHY
Patients underwent angiographic follow-up at four and nine months with preparation and percutaneous access being performed according to the standard hospital practice. Angiograms were recorded in two different orthogonal views, matching the projections at pre- and post-procedure, after intracoronary administration of nitroglycerine. For every patient, in-stent and in-segment measurements were performed by quantitative angiographic analysis (QCA). QCA analysis was conducted by an independent core laboratory (Medstar Health Research Institute, Washington, DC, USA). Quantitative measurements included minimal lumen diameter (MLD), reference vessel diameter (RVD), late luminal loss (LLL), percentage diameter stenosis (DS) and binary restenosis (BS) evaluated in-stent and in-segment. All QCA analyses were performed utilising QCA for Research CASS 5.6 software (Pie Medical Imaging, Maastricht, The Netherlands).
INTRAVASCULAR ULTRASOUND
Intravascular ultrasound was performed in a subgroup (n=15) of patients at baseline, and at four and nine months follow-up visit. All images were performed using a motorised pullback system with a pullback speed of 0.5 mm/sec. IVUS analysis was conducted by an independent core laboratory (Medstar Health Research Institute, Washington, DC, USA). The analysis was conducted utilising the Echoplaque software package version 3.0.41 software and INDEC Medical Imaging Assistant version 4.3.4C software.
STATISTICAL ANALYSIS
The study was designed to yield first data on the safety and clinical performance of the new hybrid Orsiro drug-eluting stent system and was therefore considered explorative. Resulting values will be the basis for sample size calculations for future studies. Based on similar studies with other drug-eluting stent systems, a sample size of n=30 patients was expected to be sufficient to assess the safety and clinical performance of the device. Data are presented using descriptive statistical methods. For metric data sets, the mean values, standard deviation, median, maximum and minimum and 95% confidence interval for the mean were calculated. For categorical data, absolute and relative frequencies were determined and the exact 95% confidence intervals for proportions were calculated. A p-value <0.05 was considered as statistically significant.
ROLE OF FUNDING SOURCE
The study was designed with close collaboration between the investigators and the sponsor (Biotronik AG, Buelach, Switzerland). The sponsor had no role in data interpretation or writing the final report. The corresponding author had full access to all data of the study.
Results
PATIENT CHARACTERISTICS
A group of 30 patients with de novo coronary lesions up to 22 mm in length, in vessels with a 2.5 to 3.5 mm reference diameter, and between >50% and <90% diameter stenosis were treated with the Orsiro sirolimus-eluting stent within three weeks in July 2009. The baseline clinical characteristics are presented in Table 1. The mean age of the study population was 58.1±9.8 years, with 23% of diabetic patients and 60% of male patients. This is rather a high-risk cohort for a FIM study with 73% of patients having a previous history of myocardial infarction and 37% having been enrolled and treated with a diagnosis of unstable angina. The baseline lesion characteristics are presented in Table 2. Device and procedural success was achieved with 100% in all 30 patients.
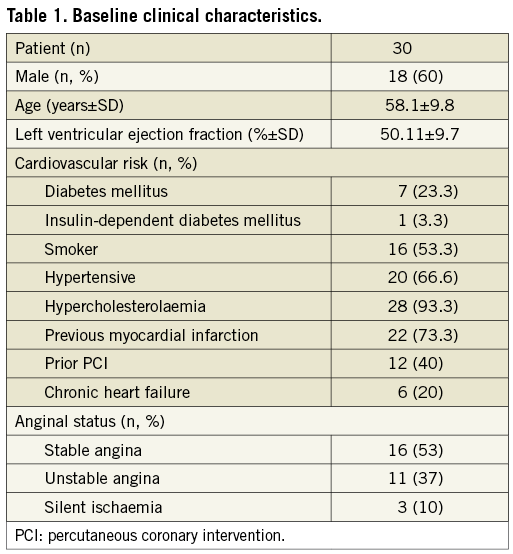
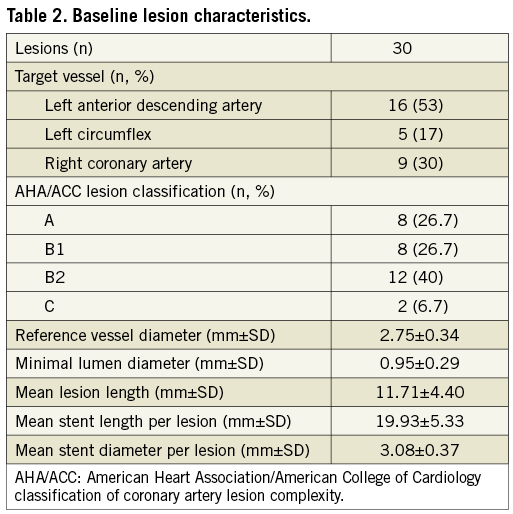
QUANTITATIVE CORONARY ANGIOGRAPHY ANALYSIS
Angiographic follow-up compliance at four and nine months was 100% in all 30 patients (Figure 1). The angiographic measurements with paired analysis of baseline, post-procedural and follow-up angiography are presented in Table 3. Angiography results of these 30 patients showed good scaffolding of the target vessel with in-stent late lumen loss at nine months of 0.05±0.22 mm. In-stent diameter stenosis after nine months was 13.60±4.27 mm with binary restenosis rate of 0%. The in-stent minimal luminal diameters before the procedure and at nine months were 0.95±0.29 mm and 2.56±0.38 mm, respectively. Figure 2A and Figure 2B demonstrate the cumulative frequency distribution curves of in-stent MLD and percentage of stenosis at pre-procedure, immediately after the index procedure, at four months and at nine months follow-up.
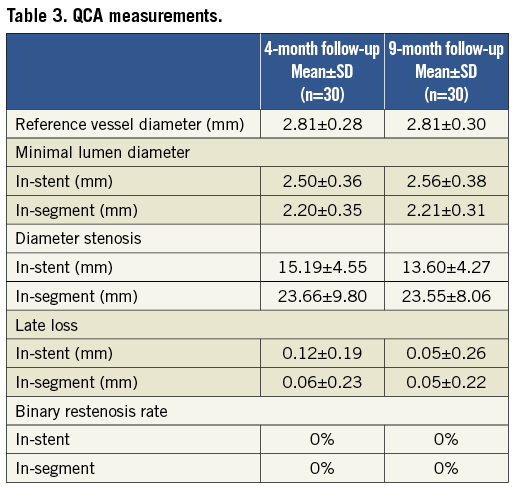
Figure 1. Flow chart of the study.
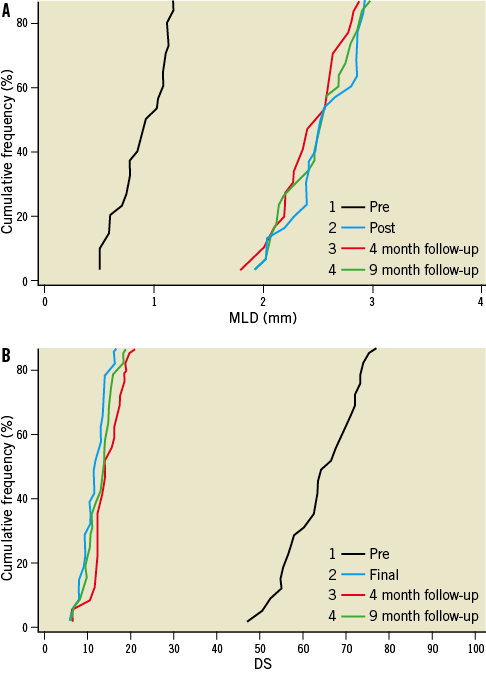
Figure 2. Cumulative frequency distribution curves of in-stent MLD and percentage of stenosis by QCA) Cumulative distribution curves for minimal luminal diameter (MLD); B) Cumulative distribution curves for diameter stenosis (DS).
CLINICAL RESULTS AND MAJOR CARDIAC EVENTS
At nine months angiographic and clinical follow-up, no death, MI or stent thrombosis was found. Clinically driven target lesion revascularisations (TLR) occurred in 2 (6.7%) out of 30 patients at the nine-month angiographic follow-up and noted to be outside of the stents but within the stented segment. One patient had a stenosis in a proximal left anterior descending artery proximal to the Orsiro stent and the second patient a stenosis in a mid-right coronary artery close to the distal part of the stent. One clinically driven target vessel revascularisation was noted at the four-month follow-up. The hierarchical MACE rate of cardiac death, MI, and clinically driven TLR at one year was 10% (3/30) (Table 4). One sudden death was noted at the one-year clinical follow-up with cause of death adjudicated as cardiac death but without MI or stent thrombosis found at the autopsy. No stent thrombosis was observed. None of the reported MACE occurred in the diabetic patients.
IVUS ANALYSIS
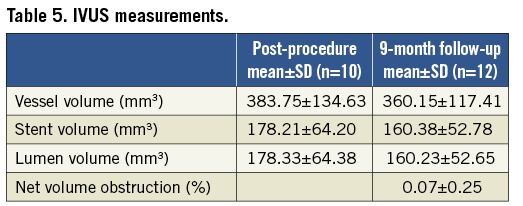
Out of the 15 patients consenting for the IVUS follow-up, paired data are available for eight patients at the four-month follow-up and nine patients at the nine-month follow-up. The remainder of the patients did not have images that could be analysed volumetrically, specifically due to technical reasons such as inconsistent IVUS pullback. The intravascular ultrasound analysis showed that the majority of the patients did not develop any neointimal hyperplasia so the mean net volume obstruction at four months follow-up was not measurable (0.0%) and extremely small with 0.07% at nine months follow-up (Table 5). There was no significant change in stent volume or lumen volume. However, the small sample size limited the power to detect small changes.
Discussion
In this prospective, first-in-man study the Orsiro sirolimus-eluting stent system showed excellent clinical performance and late lumen loss at angiographic follow-up. The patient population being complex with a high rate of diabetes and complex coronary lesions is atypical for a FIM study and provides more robustness to the efficacy of the data. This is also evinced by the lack of late catch-up in the late lumen loss values and the narrow standard deviation.
There was no myocardial infarction or stent thrombosis. There was one sudden death at one-year follow-up with cause unidentified. With the clinically driven TLR rate being 6.7%, the MACE rate of death, MI, and clinically driven TLR at one year was 10% (3/30).
In comparison, the XIENCE V® Everolimus Eluting Coronary Stent System (Abbott Vascular, Santa Clara, CA, USA), composed of a stent delivery system coated with a formulation containing the anti-restenotic drug everolimus embedded in a durable biocompatible polymer, demonstrated an angiographic in-stent late lumen loss of 0.10 mm at six months and 0.23 mm at 12 months. This was seen in the SPIRIT FIRST study, a prospective, randomised, single-blind study evaluating the XIENCE V® Everolimus Eluting Coronary Stent versus an identical Multi-Link Vision® bare metal stent in de novo lesions12. Similar to BIOFLOW-I, there were no acute or late stent thromboses reported through the one-year follow-up period13. However, the rate of major adverse cardiac events (MACE) with the XIENCE V® stent was 15.4% (4/26) at one year, compared with 21.4% (6/28) in the control arm. It is noted that, while the MACE rate in the BIOFLOW-I study was lower at 10% (3/30), the patient population was more complex in comparison to the patients in the SPIRIT FIRST study. The BIOFLOW-I patient population had higher rates of diabetes, hyperlipidaemia, current smoking status and type B2-C lesions.
There was no late catch-up of neointimal formation as evaluated by IVUS findings. There was no evidence of any toxicity in terms of aneurysm and vessel ectasia by angiogram or stent malapposition by IVUS. Pointing out the homogenous standard deviation observed at both angiographic endpoints appears far more important and strengthens the robustness of our findings regarding the performance of the Orsiro stent in preventing neointimal hyperplasia which was the main finding of this FIM study.
To be on the safe side, the dual antiplatelet therapy (DAPT) protocol in this FIM study was 12 months. Shorter durations of DAPT may apply but, since the PLLA polymer will remain for approximately 24 months, we suggest adopting the same duration of DAPT as currently used for second-generation DES.
A limitation of the study was the absence of direct comparison with other stents as seen in randomised studies. Although there were no cases of stent thrombosis or MI, the small number of patients involved in this study is limiting in the detection of adverse events and complications. This present study supports conducting randomised clinical studies to evaluate Orsiro among the other drug-eluting stents.
This third generation of DES incorporates advanced design features such as the cobalt-chromium stent platform coated with an anti-restenotic drug, sirolimus, into a biocompatible polymer with a long history of medical use. The lack of angiographic restenosis at one year demonstrated the efficacy of the stent and the hope is that such results can be consolidated in the continued follow-up. Larger cohorts comparing the Orsiro DES to second generations14 with longer-term follow-up to five years will determine if these modifications of the third-generation DES can be translated to clinical differences when compared with second-generation DES.
Appendix
Combined Clinical Events Committee & Data Safety Monitoring Board (CEC & DSMB) members: Prof. Christiaan J.M. Vrints, MD, PhD, Antwerp University Hospital, Antwerpen, Belgium; Bart J.G.L. de Smet, MD, PhD, University Medical Center Groningen, Groningen, The Netherlands; Jan Bart Hak, PhD, Xendo Drug Development BV, Groningen, The Netherlands
Conflict of interest statement
M. Hamon has received fees for lectures, advisory board or consulting services in the last two years from Cordis, Terumo, The Medicines Company, Lilly, Biotronik, Medtronic and research grants from The Medicines Company, GlaxoSmithKline and Lilly. R. Waksman serves as a consultant to Biotronik, Abbott Vascular, Boston Scientific and Medtronic Vascular. The other authors have no conflicts of interest to declare.
