- angioplasty
- transluminal percutaneous coronary
- coronary stenosis
- paclitaxel
- stents
- drug-eluting stents
Abstract
Aims: To compare the angiographic and clinical performance of a paclitaxel-eluting stent using reservoirs technology and a bioabsorbable polymer, without surface coating (CoStar), vs. an equivalent bare metal stent (BMS) using an identical metallic platform.
Methods and results: Three hundred and three (303) patients (335 lesions) with de novo coronary artery stenosis suitable for elective percutaneous treatment were randomised in an international multicentre single-blind trial to receive the CoStar stent (n=152) or the equivalent BMS (n=151). At eight months, the primary endpoint of in-segment binary restenosis was significantly lower in the CoStar than in the BMS group (17.6 vs. 30.3%, p=0.029). In-stent late loss (0.41 vs. 0.81 mm; p<0.0001) and all the other angiographic secondary endpoints also favoured CoStar. The composite of cardiac death, myocardial infarction related to the target vessel and target lesion revascularisation was significantly lower at eight months in the CoStar arm (19.7 vs. 29.1%; hazard ratio 0.54, 95% CI: 0.34-0.87; p=0.010), mainly due to lower incidence of target lesion revascularisation (15.1 vs. 26.5%; 95% CI: hazard ratio 0.45, 95% CI: 0.27-0.76; p=0.002).
Conclusions: As compared with a bare metal stent of identical design, the paclitaxel elution from reservoirs results in significantly less binary restenosis, less late loss and lower revascularisation rates at eight months. Therefore, based on these data, the CoStar paclitaxel-eluting stent was found to be effective and safe.
Introduction
Patients receiving bare metal stents (BMS) suffer from restenosis in 20.0-50.3% due to excessive neointimal proliferation1. Due to their ability to inhibit cellular proliferation, drug-eluting stents (DES) have reduced the restenosis rates to 7.9-8.9%2-5. However some reports have suggested an eventually higher incidence of late and very late stent thrombosis in DES6-10, with the common pathological finding of delayed neointimal healing and incomplete endothelialisation in fatal cases11-15. The mechanism for delayed neointimal healing and stent thrombosis seems to go beyond the antiproliferative potency of the drug and involve also other factors, like the thickness of the struts16, cracking of the polymer17, polymer-induced inflammatory reaction14,18-22 or inappropriate kinetics of drug release23,24. In some first generation DES a specific inflammatory reaction has been described, with presence of intense eosinophilic infiltrates in the vessel wall14 and in the thrombus harvested from patients suffering very late stent thrombosis19, that might be mediated by delayed type IVb hypersensitivity, recruiting preferentially eosinophils. This hypersensitivity is likely triggered by the polymer rather than by other components of the device21, given the timing of onset (later than 90 days, when the drug is no longer detectable in the vessel wall) and the presence of polymer fragments surrounded by giant cells14,22. Also inadequate pharmacokinetics of the device are known to be potentially harmful: excessive drug release during the early phase of repair might cause not only delayed healing but also toxicity, leading to smooth muscle cells necrosis, positive remodelling and acquired malapposition23.
Intense research efforts are currently aimed to optimise DES design features, to improve its safety profile and to promote complete neointimal healing, in order to prevent stent thrombosis. Reservoir technology offers considerable advantages with respect to surface polymer coating: struts are honeycombed with laser-cut holes or wells that act as drug reservoirs. This design permits precise control of the spatial drug release (abluminal/ adluminal/ bidirectional) and optimisation of the temporal elution rate using inlaid stacked layers of drug and polymer25. The polymer layers can be bioabsorbable and disappear after elution of the drug, thus circumventing the problem of delayed hypersensitivity and late inflammatory reactions associated to thrombotic phenomena. The lack of surface polymer coating avoids also the risk of cracking as previously described17, although stents with reservoirs require a specific design, with specifically engineered hinge points and bridges, to increase its flexibility and deliverability as well as preserve the structural and functional integrity of the reservoirs after the deployment stress25.
The CoStar stent (previously Conor MedSystems, Menlo Park, CA, USA, now Cordis Corporation, Warren, NJ, USA) consists of a new cobalt-chromium platform (Unistar, Conor MedSystems, Menlo Park, CA, USA) with reservoirs containing a bioabsorbable poly-(lactide-co-glycolide) (PLGA) polymer and paclitaxel at a dose of 10µg/17 mm of stent. The enhanced flexibility was achieved by a new stent design with bridge elements and ductile hinges (Figure 1). The elution of the drug is solely abluminal and prolonged to 30 days, coupled to the progressive degradation of the PLGA polymer by hydrolysis. This release formulation is the result of an evidence-based clinical selection process among other formulations, being the one with lowest incidence of major adverse cardiovascular events (MACE)26 and lowest angiographic late loss27. The thickness of the struts is 90 µm. The CoStar stent failed to prove non-inferiority vs. a first-generation surface-coating paclitaxel-eluting stent (Taxus Express; Boston Scientific, Natick, MA Grove, MN, USA) in the COSTAR-II trial28. Furthermore, the performance of the CoStar stent in this study was assumed not to be significantly different from the “imputed”, i.e. theoretically constructed, virtual BMS27. These results questioned the efficacy of reservoirs DES as drug-delivery technology. Purpose of this study was to compare the performance of the CoStar reservoirs DES vs. a BMS of identical design but with empty reservoirs.
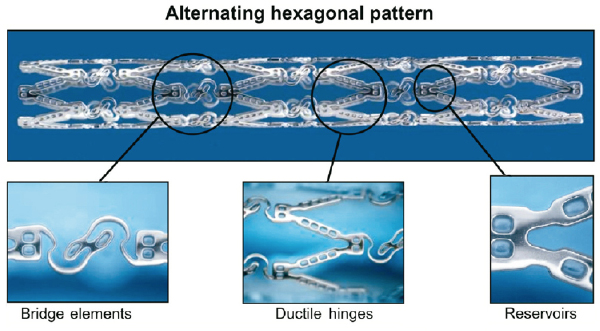
Figure 1. Design of the CoStar DES. The new cobalt-chromium platform has reservoirs containing a bioabsorbable poly-(lactide-co-glycolide) (PLGA) polymer and paclitaxel at a dose of 10 µg/17 mm of stent. Its enhanced flexibility was achieved by a new stent design with bridge elements and ductile hinges.
Methods
The EUROSTAR-II trial was an international multi-centre, randomised, single-blind trial evaluating the efficacy and safety of the CoStar paclitaxel-eluting stent with reservoir technology vs. a control of the identical BMS platform without drug or polymer (Unistar; Conor MedSystems, Menlo Park, CA, USA) for elective treatment of de novo lesions in native coronary arteries.
Study endpoints
Primary endpoint for the study was in-segment binary restenosis rate at eight months by quantitative coronary angiography (QCA). Angiographic secondary endpoints at eight months were: 1) in-stent and in-segment late lumen loss; 2) in-stent and in-segment minimal lumen diameter (MLD). Clinical secondary endpoints were: 1) MACE at 30 days and eight months, defined as an adjudicated composite of death that cannot be clearly attributed to a non-cardiac cause or non-intervention vessel, new myocardial infarction (MI, Q- or non-Q-wave) that cannot be clearly attributed to a non-intervention vessel, according to World Health Organisation criteria29 and target vessel revascularisation (TVR); 2) clinically-driven TVR; and 3) clinically-driven TLR. Combined secondary endpoints were: 1) device success, defined as attainment of <50% in-stent residual stenosis by QCA as final result of the intervention, in absence of device malfunction, and 2) procedural success, defined as attainment of <50% in-stent residual stenosis by QCA as final result of the intervention, in absence of in-hospital MACE.
Sample size calculation
This trial was designed as a superiority one-sided trial of the DES vs. the control arm using the BMS of identical design. Based on prior studies, the estimated incidence of the primary endpoint was estimated in 5% for the CoStar DES intervention arm27 and in 15% for the UniStar BMS active control arm26. On these assumptions and for a one-sided a error of 0.05, a minimum sample size of 131 patients per treatment arm was calculated to yield a greater than 80% power of finding a significant difference, using the normal method with Fleiss’ correction. Accounting for up to 10% patients lost to follow-up, the final sample size calculation resulted in 146 patients per group.
Study population
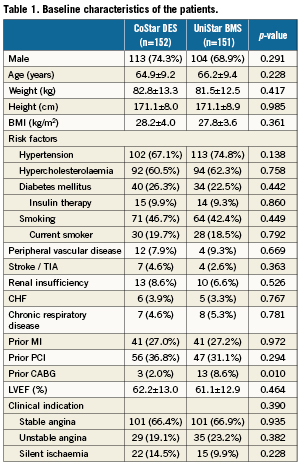
Patients between 18-80 years of age, with stable or unstable angina pectoris or with a positive functional test for ischemia and up to two discrete de novo lesions in native coronary arteries, amenable to treatment with percutaneous coronary intervention (PCI) using the study stents were enrolled into the trial. Eligible lesions had to be between 50% and 99% diameter stenosis, reference vessel diameter (RVD) 2.5-3.5 mm and length ≤25 mm by visual estimation that could be treated with a single study stent. TIMI flow pre-intervention had to be ≥I. Study lesions should not have undergone any previous interventional procedure of any kind, and no additional treatment should be planned for the patient in the following 30 days. Exclusion criteria were: cerebrovascular event or transient ischemic attack within the prior six months, percutaneous or surgical coronary revascularisation within the prior 30 days, acute myocardial infarction within the prior 72 hours, cardiogenic shock, unstable ventricular arrhythmias, left ventricular ejection fraction <30%, serum creatinine >2.5 mg/dL, known hypersensitivity to any of the components of the study devices or to the procedure medication, episode of gastrointestinal bleeding in the preceding three months, contraindication for dual antiplatelet therapy, any other clinical condition conferring the patient a life expectancy <2 years, presence of >2 lesions (or >1 lesion in the same coronary artery) requiring treatment, target lesion involving a bifurcation with a side branch >2 mm in diameter, detection of intraluminal thrombus visible in the angiography and planned used of adjunctive coronary devices (e.g., cutting-balloon or atherectomy).
All patients in the trial provided written informed consent before enrolment, and were randomly allocated on a 1:1 basis to receive the CoStar paclitaxel-eluting stent with reservoir technology or the UniStar BMS with identical, but empty reservoirs. Allocation to treatment used a random computer-generated sequence of numbers, and sequentially numbered sealed envelopes available at each study site. The patient, but not the operator, was kept blinded to the allocation. The study was conducted in accordance with Good Clinical Practice, Declaration of Helsinki and local regulations, and protocol was approved by the Ethical Committees of the centres involved in the trial.
Description of the intervention and follow-up
All patients received 100 mg of aspirin at least one hour before the intervention and a minimum loading dose of 300mg of clopidogrel prior or immediately following the procedure. Use of glycoprotein IIb/IIIa inhibitors was left at the operator’s discretion. Intravenous heparin was administered during the procedure to keep an activated clotting time ≥250 seconds, or 200-250 if a glycoprotein IIb/IIIa receptor blocker was administered.
The interventions were performed with a ≥6 Fr guiding catheter. Direct stenting or predilatation with a balloon shorter and at least 0.5 mm smaller in diameter than the study stent were both allowed. The study stents (as described above) were available at 2.5, 3.0 and 3.5mm diameter, and at 10, 16, 22, 28 and 33mm length. The implanted stent had to cover the whole target lesion length and the entire ballooned segment in case of predilatation, extending at least 2mm beyond on each side. Use of additional stents had to be avoided, except in the cases of insufficient lesion coverage or bailout procedure. If the patient required additional bailout stents, these had to be identical to the initial study stents implanted. The stent was deployed at an inflation pressure between nominal and rated burst pressure to achieve full expansion, complete apposition and a final diameter stenosis <10%. If necessary, the stent could be post-dilated with a balloon shorter than the stent length at the operator’s discretion. IVUS guidance was allowed but not mandatory. Systematic monitoring of ECG and cardiac serum markers was performed in all patients after the procedure and before discharge.
After the intervention, patients were kept on dual antiplatelet therapy with 100 mg of aspirin and 75 mg of clopidogrel daily for a minimum of six months, followed by daily aspirin indefinitely. Clinical follow-up visits were scheduled 30 days and eight months post-procedure, and angiographic follow-up at eight months.
Quantitative coronary angiography (QCA) analysis
Coronary angiography was performed according to standard procedures30. QCA analysis was performed with the CAAS II system31 (Pie Medical BV, Maastricht, The Netherlands) in a core-lab setting (Bio-Imaging Technologies, Leiden, The Netherlands) by analysts blinded to patients’ characteristics and to the allocation to treatment. The analysis results were reported for the stented segment (in-stent) and for the segment comprising 5 mm proximal and distal to the stent edges (in-segment). MLD was automatically detected by the software. RVD at the point of MLD was calculated by the software by interpolation. % diameter stenosis was calculated as: (1-[MLD/RVD])*100. Binary restenosis was defined as% diameter stenosis ≥50%. In-stent and in-segment late lumen loss were defined as the difference between MLD at eight months follow-up and the respective post-procedure MLD.
Statistical analysis
Results are reported as mean±standard deviation for continuous variables, and as count (percent) for nominal variables. Continuous variables were compared with Fisher’s t-test for independent samples. Nominal variables were compared with Pearson’s chi-square, or Fisher’s exact test if the expected frequency was <5 in any cell.
Clinical and safety endpoints followed a hierarchical events model. Incidences of the different endpoints at 30 days were calculated and compared as risk ratios. Results at eight months were analysed as events-free survival using Cox proportional hazards regression and log rank tests.
All statistical analyses were performed according to the intention-to-treat principle, using the PASW 17.0.2 statistical package (SPSS Inc., Chicago, IL, USA).
Results
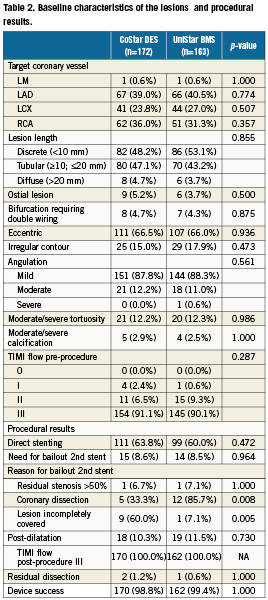
Three hundred and three (303) patients (335 lesions) were enrolled in the EUROSTAR-II trial at 18 different European sites: 152 in the CoStar DES group, and 151 in the Unistar BMS group (Figure 2). Table1 and Table2 show the baseline characteristics of patients and lesions, respectively, with no significant difference in any of the variables tested, except for a larger proportion of prior coronary artery bypass graft in the UniStar group (p=0.010). QCA analysis did not show significant differences in the pre-procedural analysis of the lesions (Table 3). Both groups were also comparable with respect to QCA results post-stenting, except for a slightly higher residual diameter stenosis in the CoStar than in the UniStar subgroup (21.15 vs. 18.62%, respectively; p=0.030).
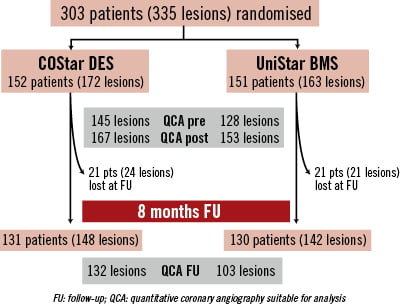
Figure 2. Flow chart of the study.
Forty-two (42) patients (13.9%) were lost for angiographic follow-up: 21 (13.8%) and 21 (13.9%) in the CoStar and UniStar groups, respectively. Clinical follow-up was completed in all patients at eight months. Median FU time was 243 days, inter-quartile range (217-250 days). The primary endpoint (in-segment % binary restenosis) was significantly reduced in the Costar arm: 17.6 vs. 30.3%, p=0.029; (Table 3, Figure 3). Significant differences in favour of CoStar were also found in all the angiographic secondary endpoints (Table 3, Figures 3-5).
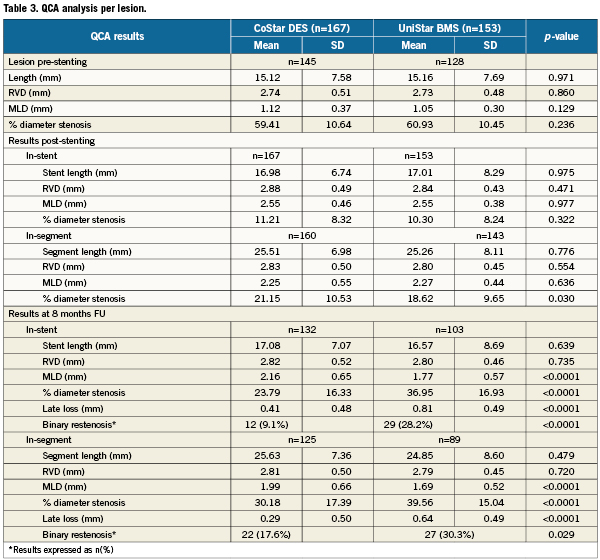
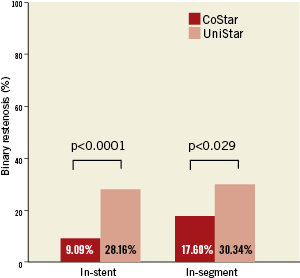
Figure 3. In-stent and in-segment binary restenosis (primary endpoint) of the CoStar DES and the UniStar BMS at eight months follow-up.
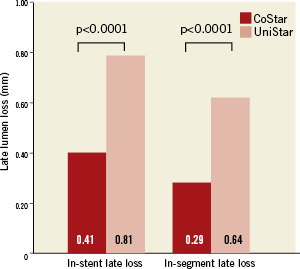
Figure 4. In-stent and in-segment late lumen loss of the CoStar DES and the UniStar BMS at eight months follow-up.
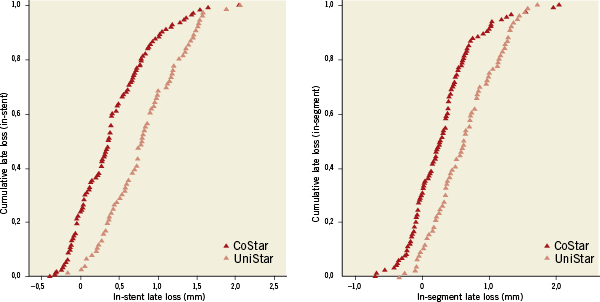
Figure 5. In-stent and in-segment late lumen loss cumulative curves of the CoStar DES and the UniStar BMS at 8 months follow-up.
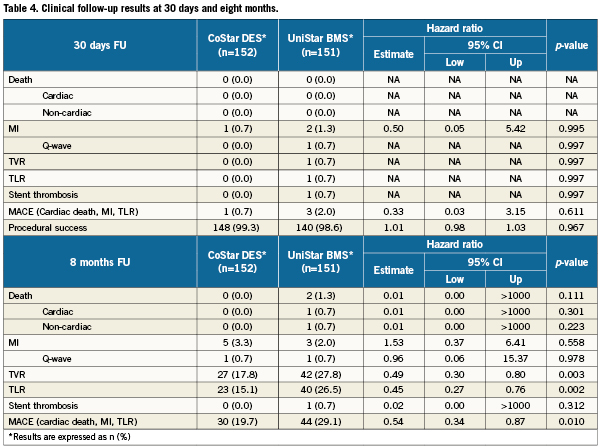
Regarding the clinical and safety endpoints, no significant difference was found between groups at 30 days (Table 4). However, the incidence of MACE was significantly reduced at eight months in the CoStar arm (19.7 vs. 29.1%; hazard ratio 0.54, 95% CI: 0.34-0.87; p=0.010). Similar death, and MI rates were found at eight months in both treatment groups, but the incidence of TLR was significantly lower in CoStar (15.1 vs. 26.5%; hazard ratio 0.45, 95% CI: 0.27-0.76; p=0.002). A single case of stent thrombosis was registered in the UniStar group seven days after the intervention (subacute), and classified as definite according to ARC criteria32.
Discussion
The results of this EUROSTAR-II trial prove the efficacy of reservoirs technology for inhibition of neointimal hyperplasia and clinically relevant prevention of restenosis, compared to an identical BMS platform. The primary endpoint (in-segment % binary restenosis) was significantly lower in the group treated with a CoStar DES than in the group treated with the UniStar BMS. Other secondary endpoints addressing the inhibition of neointimal hyperplasia and prevention of restenosis, like late loss, or incidence of TVR and TLR, were also significantly in favour of the reservoirs DES. The reservoirs DES also proved to be superior in secondary clinical endpoints, like the incidence of the composite of death, MI and TLR, although this clinical superiority was mainly due to the reduction of TLR, showing similar rates of death and MI. This finding is consistent with the angiographic findings, and can be interpreted as efficient and clinically relevant prevention of restenosis, without clinical safety concerns.
The results of the COSTAR-II study had questioned the efficacy of reservoirs DES28: the reservoir paclitaxel-eluting CoStar stent failed to prove non-inferiority vs. a first-generation surface-coating paclitaxel-eluting stent (Taxus Express, Boston Scientific, Natick, MA, USA). Furthermore, in the COSTAR-II study, the performance of the CoStar reservoirs DES was assumed not to be significantly different from the “imputed”, i.e., theoretically constructed, virtual BMS28. The hereby reported EUROSTAR-II trial was run simultaneously to the COSTAR-II trial. In contrast to COSTAR-II, EUROSTAR-II is the only randomised trial directly comparing the performance of the same reservoirs DES vs. an equivalent BMS platform of identical design. The EUROSTAR-II results definitely answer the question about the efficacy of reservoirs DES vs. BMS, at a higher level of evidence than indirect hypothetical placebo imputations of COSTAR-II. Our results are also more consistent with preceding evidence about the CoStar stent25-27 and other reservoirs DES33. The hereby reported angiographic results for the CoStar DES (in-stent binary restenosis 9.1%, in-stent late loss 0.41 mm) are in between the ones obtained in the CoStar-II trial (17.9%, 0.64 mm, respectively)28 and the values from preceding studies with the same device (0-5.7%, 0.28-0.38 mm)26,27; being similar to Taxus Express in its pivotal trial (5.5%, 0.39 mm)5. The angiographic results for the UniStar BMS are also comparable to those reported for the BMS in TAXUS IV5. Putting into perspective the results of EUROSTAR-II with the preceding results, it seems that the first studies about the reservoirs CoStar DES overestimated its efficacy26,27, but the present study proves that the reservoirs paclitaxel-eluting CoStar DES prevents restenosis compared to an equivalent BMS.
The incidence of MACE in this trial is however much higher than in any preceding study5,26-28, so for the CoStar DES group as for the BMS control group. This excess of MACE is exclusively due to a much higher incidence of revascularisation: TVR for the CoStar DES was 17.8%, whereas it was 8.1% in the COSTAR-II28; 2.8% in EUROSTAR-I27 and 2.6% in PISCES26. Revascularisation in the BMS group was also higher than in prior studies: TVR for the UniStar BMS was 27.8%, whilst it was 12.0% in the BMS arm of the TAXUS IV trial5. The reason explaining this excess of revascularisation and consequently of MACE can be the coincidence in time of the clinical and angiographic follow-up at eight months, resulting in some “oculostenotic” revascularisations performed during routine angiographic follow-up and accounted as clinically-driven. In fact the curves in Figure 6 show a steep increase in both TLR and composite MACE around 244 days (eight months). In contrast, in COSTAR-II the primary clinical endpoint could not have been affected by the “oculostenotic” revascularisation because it was defined at eight months with angiographic follow-up at nine months. However the coincidence in time of the angiographic and clinical follow-up explains only partially these results: as compared with the TAXUS IV trial, binary restenosis was twice bigger in the CoStar than in the paclitaxel-surface coated Taxus Express stent, even though their late loss was similar and the restenosis rate in the BMS control arms was comparable5. Thus, the CoStar stent might be less efficient than Taxus for inhibition of neointimal hyperplasia, as suggested by COSTAR-II28. An optimised design of the honeycombed stent platform, and the different anti-proliferative drugs, with different dosage and kinetics of release, could have contributed to improve the clinical and angiographic outcomes of DES reservoir technology, as recently reported33.
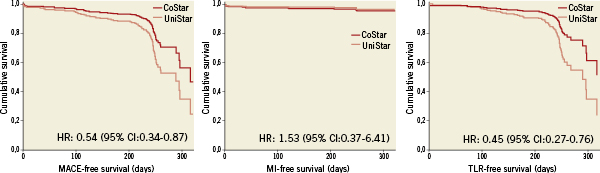
Figure 6. Event-free survival plots for the composite endpoint of major adverse cardiovascular events (MACE) at eight months, comprising cardiac death, myocardial infarction (MI) and clinically-driven target lesion revascularisation (TLR).
Limitations
This trial was performed on a selected population with respect to clinical and angiographic features. This must be taken into account in the interpretation and generalisation of the results.
Although the randomisation process worked well in general, it resulted in the imbalanced distribution of the variable “prior coronary artery bypass graft surgery” between treatment groups. This imbalance might have biased the results at some extent, but the magnitude of this bias was deemed minor and therefore an eventual modification of the pre-specified statistical analysis was not considered to be justified.
Loss at angiographic follow-up was approximately 14%, therefore it remained in the range considered acceptable for the validity of studies with a primary angiographic endpoint. The attrition at follow-up did not seem to affect selectively to any of the treatment groups. Nonetheless, some angiographic studies were discarded for QCA analysis due to insufficient quality. This might have introduced some selection bias in the results, although it affected both groups alike. In spite of this limitation, the QCA results are consistent with the clinical efficacy variables, less affected by loss or selection.
Although the absence of thrombotic events in the CoStar DES group is compatible with the hypothesis that a bioabsorbable polymer might avoid delayed hypersensitivity reactions triggering very late thrombosis, this study, like all other DES randomised trials published so far, is underpowered for testing stent thrombosis and no valid conclusion can be stated in this regard.
Conclusion
As compared with an equivalent bare metal stent, paclitaxel elution from reservoirs resulted in significantly less binary restenosis, less late loss and lower revascularisation rates at eight months. No safety concerns were observed.
Funding
This study was enabled by a study grant of BIOTRONIK, Berlin, Germany. The Steering Committee comprised Sigmund Silber, MD (principal investigator, Munich, Germany) as well as Harry Suryapranata, MD (Zwolle, The Netherlands) and Bernard Chevalier, MD (Massy, France). CRO and independent external monitoring was performed by DATATRAK, Bonn Germany. Data and Safety Monitoring Committee (DMSC) / Clinical Events Committee (CEC) members were Marcus Lins, MD, (Kiel, Germany), Didier Blanchard, MD, (Tours, France) and Jan Bart Hak, PhD, (Chairman, Groningen, The Netherlands).
Conflict of interest statement
The co-authors (except J.L. Gutiérrez-Chico and P.W. Serruys) received a research grant for this study from Biotronik, Berlin, Germany. No other possible conflicts of interests regarding this study were reported.
