![]()
Abstract
Aims: The DIRECT study is a first-in-human evaluation of the safety and efficacy of the Svelte sirolimus-eluting coronary stent mounted on a fixed-wire, “all-in-one” integrated delivery system (IDS) in patients with de novo coronary artery lesions. The system permits easy delivery, deployment and post-dilatation of a cobalt-chromium stent eluting sirolimus from a fully bioabsorbable amino acid coating. The stent on its IDS has a very low profile, and is designed specifically to facilitate direct stenting.
Methods and results: Patients with symptomatic ischaemic heart disease and a single de novo native coronary lesion suitable for percutaneous coronary intervention were prospectively enrolled at four New Zealand sites. The lesion length had to be <23 mm and the vessel reference diameter 2.5-3.5 mm. The primary safety and efficacy endpoints were target vessel failure (TVF) and angiographic in-stent late lumen loss (LLL) at six months, respectively. Twenty-nine of 30 enrolled patients completed six-month follow-up. TVF occurred in two patients (7%). The in-stent LLL was 0.22±0.27 mm. No patient had clinically-driven target lesion revascularisation. Intravascular ultrasound neointimal volume was 3.3±4.4 mm3 and volume obstruction was 2.7±4.5% at six months. Optical coherence tomography showed 98±4% strut coverage at a depth of 0.12±0.06 mm. No patient developed stent thrombosis.
Conclusions: Percutaneous coronary intervention using the Svelte sirolimus-eluting coronary stent mounted on an IDS appears safe and effective in de novo coronary artery lesions, with minimal in-stent proliferation and excellent stent strut coverage at six months.
Background
Drug-eluting stents (DES) have evolved to become the standard of care for patients undergoing percutaneous coronary intervention (PCI). Early DES dramatically reduced the in-stent neointimal hyperplasia seen with their bare metal equivalents. The markedly reduced rates of angiographic restenosis were mirrored by impressive reductions in the need for clinically-driven target vessel revascularisation (TVR)1-3. Rapid growth in the DES market was transiently halted by concern regarding the infrequent occurrence of late stent thrombosis, a problem which has lessened with current third-generation stents. DES technology continues to evolve. Apart from improving existing stent platforms, a recent focus has been on developing more biocompatible polymers, to reduce further the risk of late stent thrombosis.
Making stent delivery and deployment easier could improve procedural success and reduce periprocedural complications. One strategy is to facilitate the direct stenting technique in a subset of patients. Suitable lesions for direct stenting include those with minimal calcification in which an accurate assessment of reference vessel diameter and lesion length is possible without pre-dilatation. Direct stenting may reduce distal vessel embolisation and no-reflow, and has been associated with a trend towards improved clinical outcomes4. Other potential advantages include a shorter procedure time, less radiation exposure and lower procedural cost5-9.
The Svelte (Svelte Medical Systems, New Providence, NJ, USA) integrated delivery system (IDS) employs an ultra-low-profile, cobalt-chromium stent platform developed to facilitate direct stenting. A fully bioabsorbable amino acid drug carrier eluting sirolimus was added to this platform. The DIRECT (Direct Implantation of a Rapamycin-Eluting stent with bioabsorbable Carrier Technology) study was designed to assess the clinical safety and efficacy of this device for treatment of de novo stenoses in native coronary arteries.
Methods
DEVICE DESCRIPTION
The Svelte drug-eluting coronary stent IDS is a proprietary low-profile drug-eluting stent mounted on a fixed-wire delivery system (Figure 1). It is designed specifically for direct stenting and is 5 Fr guide catheter-compatible10,11 . The stent comprises a thin-strut (81 µm) cobalt-chromium alloy L-605 stent coated with a thin (6 µm), fully bioabsorbable drug carrier (DSM, Heerlen, The Netherlands) eluting the well-studied compound sirolimus (220 µg/cm2). The drug carrier is composed of naturally occurring amino acids. Extensive preclinical data were collected using porcine models. Specifically, 49 animals were implanted with 126 stents to assess pharmacokinetics (drug elution rates and drug tissue concentration levels), drug blood levels, drug carrier degradation (duration and uniformity), tissue response and general safety parameters (endothelial impairment, inflammatory and hyperplastic response) at various time points up to 390 days (Figure 2). Blood, tissue, scanning electron micrography, histopathology and histomorphometry testing were performed. The amino acids were shown to dissolve in a predictable and uniform manner via enzymatic surface erosion, with complete dissolution achieved over approximately 12 months from baseline. Elution of the drug follows classic unidirectional (Fickian) diffusion kinetics. Drug tissue concentration and fractional release is similar to previously studied stents eluting sirolimus12 and everolimus13. In other preclinical models, the Svelte drug carrier appeared to have good mechanical integrity and be highly biocompatibile. The absence of both significant pH change and activation of the complement cycle seems to mitigate the potential for a hyperplastic inflammatory response, with minimal neointimal hyperplasia through 390 days found in the porcine model.
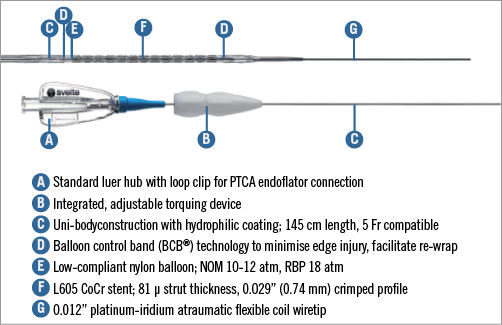
Figure 1. The Svelte sirolimus-eluting stent and IDS.
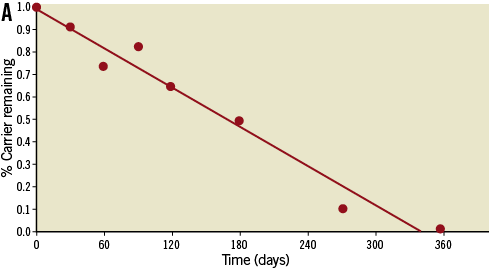
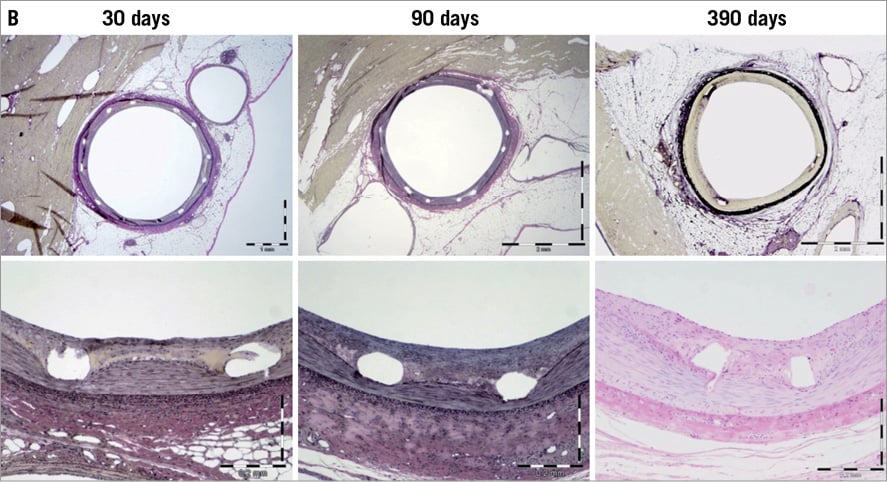
Figure 2. A) Loss in drug carrier weight from porcine explanted stents demonstrates complete erosion from the stent surface within 1 year. B) Representative porcine coronary images showing mitigation of the neointimal hyperplastic and inflammatory response through 390 days.
The IDS consists of a single-lumen catheter with a 0.012” diameter platinum-iridium flexible coil wire tip 22 mm in length which is manually shaped in a standard manner. The delivery system incorporates proprietary elastic balloon control bands (BCBs) located on the proximal and distal balloon shoulders to focus pressure inside the stent, providing controlled deployment with minimal balloon expansion beyond the stent edges (Figure 3). The stent delivery balloon is low-compliant, increasing in diameter by approximately 0.25 mm at the balloon rated burst pressure (RBP) of 18 atm, to allow high-pressure post-dilatation with the delivery system. An integrated, adjustable torquing device located on the proximal end of the catheter shaft enables the IDS to be navigated through the vasculature in a similar manner to manipulating a guidewire.
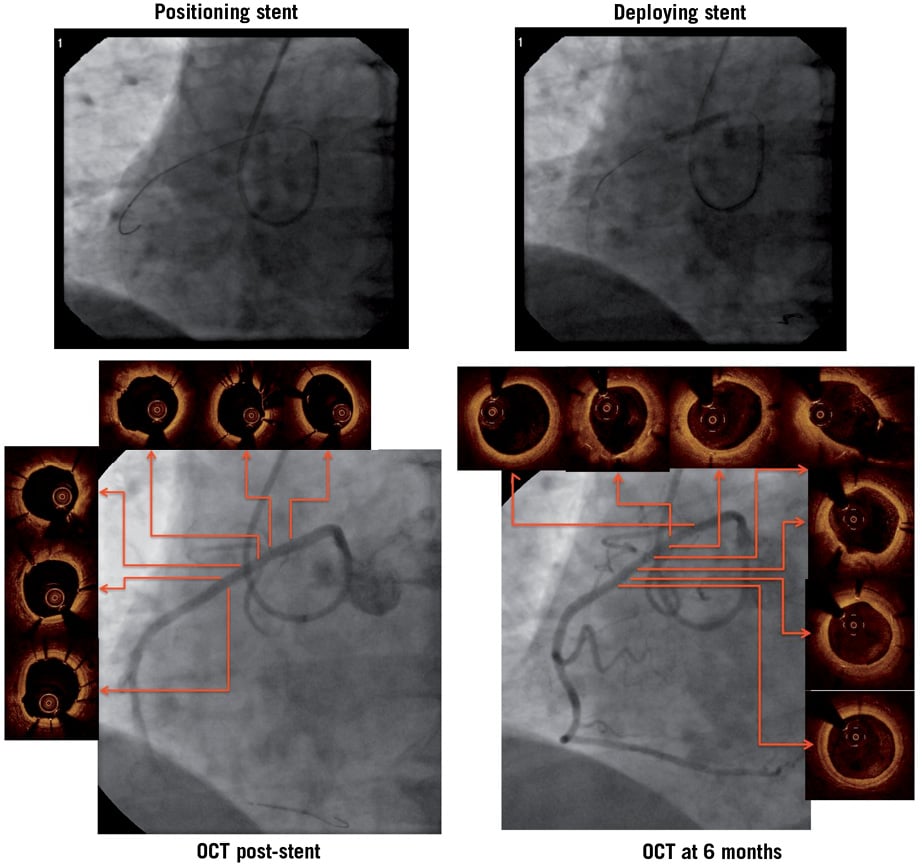
Figure 3. Angiographic and OCT images from a patient with a lesion in the mid portion of an anomalous right coronary artery. The vessel arose from the anterior aspect of the aorta at an acute angle, affording very limited guide support.
STUDY OVERVIEW AND PATIENT POPULATION
The DIRECT study was a prospective, open-label, single-arm, multicentre study designed to evaluate the safety and efficacy of the Svelte sirolimus-eluting coronary stent mounted on an IDS. Patients aged >18 years of age with symptomatic ischaemic heart disease due to a single de novo stenotic lesion within a native coronary artery suitable for percutaneous coronary intervention were eligible for inclusion in the study. The lesion length had to be <23 mm and vessel reference diameter between 2.5 and 3.5 mm. Inclusion of one non-target lesion was permitted, provided that lesion was first successfully treated with a commercially approved stent. In patients with ST-segment elevation myocardial infarction (STEMI), the culprit lesion must have been successfully treated and the patient must have remained haemodynamically stable for a minimum of 72 hours prior to enrolment. Patients with non-ST-segment elevation myocardial infarction (NSTEMI) could be included if the plasma creatine kinase (CK) was normal within 24 hours prior to the procedure.
Patients with angiographic evidence of thrombus, total occlusions (TIMI 0/1 flow), unprotected left main stem lesions, severe proximal vessel tortuosity, significant calcification, ostial lesions or lesions involving a side branch >2.0 mm were excluded from treatment with the study stent. Other exclusion criteria included active enrolment in another clinical trial, treatment with a DES in the target vessel within the preceding nine months or BMS within the preceding six months, or prior stent placement within 10 mm of the target lesion.
The study was approved by the independent Research Ethics Review Committee (Ministry of Health, New Zealand), and conformed to principles of good clinical practice and the Declaration of Helsinki. All patients provided written, informed consent.
INTERVENTIONAL PROCEDURE
All patients received aspirin (minimum 75 mg daily, commencing at least 24 hours prior to the procedure and continued indefinitely) and clopidogrel (minimum 600 mg loading dose within 24 hours prior to the procedure and then 75 mg daily for a minimum of six months). Choice of vascular access (i.e., radial or femoral) was left to operator discretion. During the procedure, either heparin or bivalirudin, with or without the addition of a glycoprotein IIb/IIIa inhibitor, was administered as per operator preference. Monitoring of activated coagulation time (ACT) was not mandatory.
The stent sizes available for the study were 2.5, 3.0 and 3.5 mm in diameter and 18 and 23 mm in length. Baseline coronary angiography was performed in at least two orthogonal views free of vessel overlap or foreshortening, following 100-200 mcg of intracoronary nitroglycerine. While direct stenting was preferred, both pre- and post-dilatation were permissible, at operator discretion.
Following stent deployment (±post-dilatation with the Svelte IDS or another balloon), coronary angiography using the same views was repeated after 100-200 mcg of intracoronary nitroglycerine was administered. All subjects were scheduled to undergo intravascular ultrasound (IVUS) evaluation (iLab; Boston Scientific, Natick, MA, USA). Optical coherence tomography (OCT, C7; St. Jude Medical, Minneapolis, MN, USA) was performed in patients treated at centres with this imaging modality. All intracoronary imaging included at least 5 mm of vessel both distal and proximal to the study stent.
PATIENT FOLLOW-UP
All patients were scheduled to undergo clinical follow-up at 30 days, six months and annually for five years from the index procedure. Repeat coronary angiography with QCA and IVUS evaluation was performed at six months. OCT was repeated in those patients treated at centres with this imaging modality.
Study endpoints and definitions
Study endpoints used Academic Research Consortium (ARC) definitions14. All study safety events and endpoints were reviewed and adjudicated by an independent joint data safety monitoring board (DSMB) and clinical event committee (CEC).
The primary safety endpoint was angiographic target vessel failure (TVF) at six months. This was defined as a composite of cardiac death, target vessel MI (Q or non-Q-wave), or clinically driven target vessel revascularisation (TVR). The primary efficacy endpoint was angiographic in-stent late lumen loss (LLL) at six months, defined as the difference in minimal lumen diameter (MLD) post index procedure and at follow-up as measured by quantitative coronary angiography (QCA) using the CASS-5 analysis system (Pie Medical, Maastricht, The Netherlands).
Secondary efficacy endpoints included acute success, defined as device (attainment of <30% final residual stenosis in the target lesion using the study stent), lesion (attainment of <30% final residual stenosis in the target lesion using a study or non-study stent) and procedure success (lesion success and absence of in-hospital MACE); cardiac death, defined as any death due to a proximate cardiac cause; myocardial infarction (MI); revascularisation, defined as a composite of target lesion revascularisation (TLR), target vessel revascularisation (TVR) and revascularisation of a non-target vessel, reported at 30 days and six months; and stent thrombosis (ST).
MACE was defined as all-cause mortality, MI or repeat revascularisation (including non-target vessel). MI was defined as: a) Q-wave MI –chest pain or other acute symptoms consistent with myocardial ischaemia and new pathological Q-waves in two or more contiguous ECG leads (in the absence of timely cardiac enzyme data), or new pathologic Q-waves in two or more contiguous ECG leads and elevation of cardiac enzymes above normal; b) non-Q-wave MI (NQWMI) – elevation of post-procedure CK ≥3x the upper limit of normal in the presence of an elevated MB isoenzyme.
Stent thrombosis was diagnosed as definite, probable or possible using ARC definitions, and categorised by time post-stent implantation as acute (<24 hours), subacute (>24 hours to 30 days), late (>30 days to one year) or very late (>1 year). Binary restenosis was defined as lumen diameter stenosis >50% at follow-up.
All deaths were considered cardiac unless an unequivocal non-cardiac cause was established. Any unexpected death, even in subjects with co-existing potentially fatal non-cardiac disease (e.g., malignancy, infection), was classified as cardiac.
SAMPLE SIZE AND STATISTICAL ANALYSIS
The DIRECT study was designed to provide adequate preliminary data about the performance, feasibility and safety of the Svelte drug-eluting coronary stent mounted on an IDS, while minimising the number of subjects exposed to a novel technology. The sample size of 30 was deemed sufficient to meet these objectives.
Discrete variables are expressed as frequencies and percentages with continuous data expressed as mean and standard deviation or median and range for parametric and non-parametric data, respectively. All statistical analyses were performed using SAS (version 6.12 or higher; SAS Institute Inc., Cary, NC, USA) or S-Plus (version 4.0 or higher; SAS Institute Inc.).
DATA COLLECTION AND CORE LABORATORY
Conduct of the trial was monitored by the Pacific Clinical Research Group. An independent DSMB/CEC reviewed all adverse events independently of the study sponsor. All electrocardiograms, coronary angiograms, IVUS and OCT images were submitted to an independent core laboratory (Cardialysis, Rotterdam, The Netherlands) for review and adjudication.
Results
Thirty patients were enrolled at four clinical sites in New Zealand between November 2011 and May 2012. They were aged 61±11 years, and 24 (80%) were male. The baseline demographic data are outlined in Table 1. Fifteen (50%) of 30 lesions were B2 according to the American College of Cardiology/American Heart Association classification scheme, including one true bifurcation lesion.
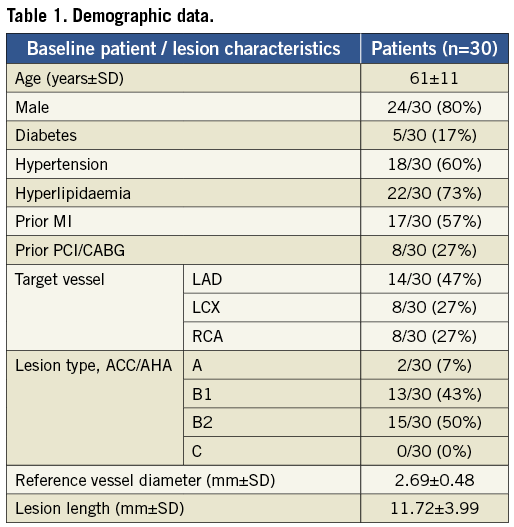
The mean lesion length, as assessed by the independent core lab, was 11.7±4.0 mm and the mean reference vessel diameter was 2.7±0.5 mm. Fourteen lesions (47%) were in the left anterior descending artery, followed by eight each (27%) in the right coronary and the left circumflex arteries, respectively. Direct stenting was performed in 23 (77%). The device, lesion and procedure success rates were 97%, 100% and 100%, respectively. The study device was not deployed in one patient, who received a non-study stent. This patient did not suffer an adverse periprocedural clinical event, and was excluded from subsequent follow-up.
QUANTITATIVE CORONARY ANGIOGRAPHY (QCA)
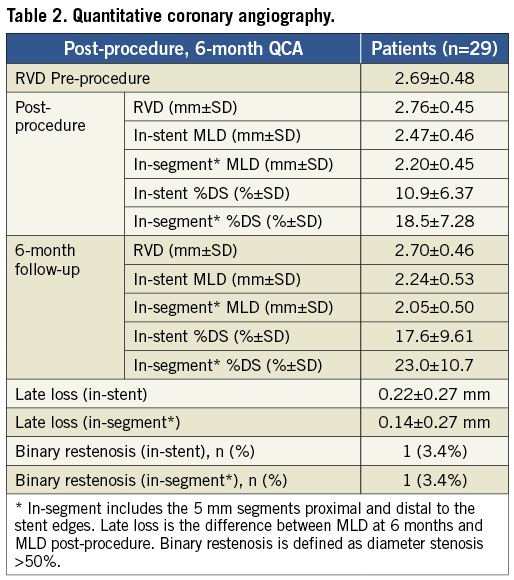
Pre, post-procedural and six-month QCA measurements were performed on all 29 patients receiving the study stent. The primary efficacy endpoint of mean in-stent LLL at six months was 0.22±0.27 mm, while the mean in-segment LLL was 0.14±0.27 mm. The percent diameter in-stent restenosis was 18±10%; one patient (3%) developed binary restenosis (Table 2).
INTRAVASCULAR ULTRASOUND (IVUS)
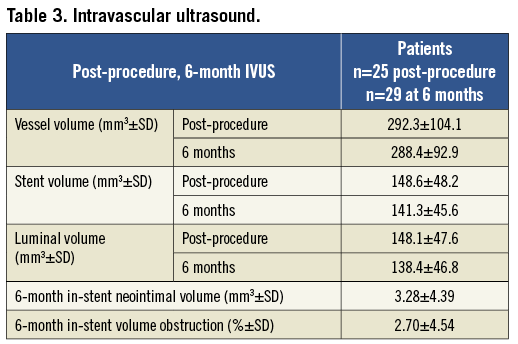
Six-month IVUS analysis was available in 28 of the 29 patients who received the study stent. In one case, malfunction of the IVUS console prevented acquisition of data at the time of angiographic follow-up. The mean in-stent neointimal volume was 3.3±4.4 mm3 and mean in-stent volume obstruction was 2.7±4.5% (Table 3).
OPTICAL COHERENCE TOMOGRAPHY (OCT)
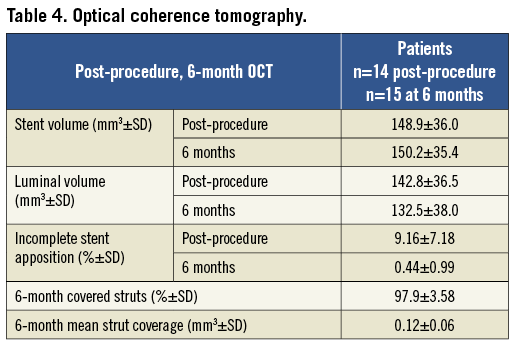
Six-month OCT analysis was available in 15 of the 29 patients who received the study stent. The mean strut coverage was 0.12±0.06 mm, with 98±4% of stent struts covered (Table 4). There was no acquired or persisting stent malapposition observed at six months.
CLINICAL OUTCOMES
Clinical follow-up was completed in all 29 patients receiving a study stent. Procedural and 30-day MACE was 0%. At six months, no patient had clinically driven TLR. The overall MACE rate of 7% was due to TVF in two patients. One asymptomatic patient had angiographically driven TLR to a stenosis at six-month follow-up (at the distal end of a study stent, treated with placement of an additional, overlapping stent). A second patient experienced angiographically driven TVR (non-TLR) to disease progression proximal to the study stent, also at the six-month follow-up visit. No patient suffered death, myocardial infarction or stent thrombosis.
Discussion
This study is the first to assess the Svelte drug-eluting coronary stent mounted on an IDS, a novel advance in stent and bioabsorbable coating technology combined with a unique delivery system specifically designed to facilitate direct stenting. None of the operators participating in this study had prior experience using the IDS. Some of the cases were challenging, such as the one depicted in Figure 3. Despite such difficulties, the high procedural success rate indicates that the device is both deliverable and user-friendly, requiring less catheter back-up support than might be expected with a fixed-wire device.
One notable feature of the IDS is the balloon technology, which combines a less compliant, thicker durometer balloon material with proprietary elastic balloon control bands (BCBs). The latter feature is designed to envelope the balloon shoulders to limit longitudinal expansion beyond the stent edges during deployment. This permits the delivery balloon to be utilised for further high-pressure post-dilatation with minimal risk of edge dissection. The increase in balloon diameter of only approximately 0.25 mm at the RBP (18 atm) may lessen the need for a separate post-dilation balloon in some patients.
The crimped cross-sectional area of the Svelte stent mounted on its fixed-wire platform is approximately one half of that of current generation DES. The very low profile of the IDS should facilitate crossing tight stenoses and increase the proportion of lesions which can be stented directly. Direct stenting has been shown to reduce contrast load, radiation exposure, procedure time and ancillary product use (guidewires, pre and post-dilatation balloons)5-10. These benefits have been shown to accrue with the bare metal version of the Svelte stent11. The low device profile also means that PCI can be readily undertaken through 5 Fr guide catheters, thereby allowing radial access even in smaller women (all procedures were via the radial approach in this study), and reducing the risk of access site complications if femoral access is used. It is even possible to deploy the bare metal version of the stent through 5 Fr diagnostic catheters10.
The IDS is suited for stenting one lesion per vessel. When there are multiple lesions in a vessel, introducing multiple IDS is more challenging and time-consuming than using standard “over-a-guidewire” techniques. Similarly, pre-planned or the likely need for IVUS or OCT, or other adjunctive guidewire-based therapies, such as thrombectomy, are better done using a conventional approach. It should be noted that, in the event of vessel dissection or occlusion, keeping the IDS in the vessel maintains lumen integrity until a buddy guidewire or second IDS can be advanced into position.
The minimal in-stent proliferation by angiography, which was confirmed by IVUS, along with excellent strut coverage by OCT and no episodes of stent thrombosis suggest that delivering sirolimus via a fully bioabsorbable, amino acid-based drug carrier warrants further evaluation. For comparison, the in-stent neointimal volume is less than half that seen with the first-in-human studies of current generation DES15,16. This novel approach to drug delivery may lessen the potential side effects of impaired endothelial recovery, fibrin deposition, burst drug release and luminal stent strut drug residue sometimes associated with durable polymer technologies17. Preclinical data demonstrated that most of the sirolimus eluted by the Svelte DES is delivered to the vessel wall within two months of stent implantation. Both sirolimus and the amino acid coating are fully absorbed within 12 months, leaving behind a thin-strut CoCr stent. In addition, other preclinical data comparing the Svelte DES to its BMS counterpart have demonstrated minimal neointimal growth and inflammation up to one year. Whether the duration of dual antiplatelet therapy can be reduced remains to be tested.
During the study there were two instances of TVF, both relating to angiography-driven TVR at six months in asymptomatic individuals. In one instance this was due to de novo disease upstream from the index lesion and, in the other case, an overlapping non-study stent was inserted to treat restenosis within 5 mm of the study stent edge. It is not certain whether this was due to geographic miss during initial stent deployment or post-dilation, or whether post-dilatation had caused vessel disruption adjacent to the stent edge (the IDS was not used for this purpose in this case).
Study limitations
Firstly, this was a small, non-randomised study intended to provide preliminary insights into the safety and efficacy of the Svelte drug-eluting IDS. Secondly, six-month clinical and angiographic follow-up may not reflect peak hyperplastic response following DES implantation. So-called “late catch-up” has been seen with some DES between six months and two years post-procedure. While the preclinical data suggest that delayed in-stent proliferation is unlikely with the Svelte platform, drug and coating, this needs to be confirmed in a larger randomised, controlled study with appropriate follow-up, which is currently underway. Finally, because there was no comparator group undergoing PCI with standard equipment, quantitation of the potential procedure time, radiation and cost savings is not possible.
Conclusion
The Svelte drug-eluting IDS appears to be an attractive option for DES deployment in selected patients with coronary disease. No clinically-driven TLR or TVR and the excellent angiographic outcomes to six months suggest that the combination of platform, drug and bioresorbable coating may well be associated with maintained longer-term safety and efficacy. Further evaluation is warranted.
Acknowledgements
The authors wish to thank their interventional cardiology colleagues, cardiac catheterisation laboratory staff, and the trial research coordinators for their contributions to this study.
Funding
Svelte Medical Systems Inc., New Providence, NJ, USA.
Conflict of interest statement
J. Ormiston serves as an advisory board member for Abbott Vascular and Boston Scientific. T. Watson has received a minor honorarium from Svelte Medical. The other authors have no conflicts of interest to declare.
