Abstract
Aims: The Svelte Stent Integrated Delivery System (IDS) is a novel fixed-wire thin-strut cobalt-chromium stent characterised by a very low entry profile. The aim of the present study is to evaluate the safety and the feasibility of the Svelte stent IDS implantation in humans.
Methods and results: The present investigation is a prospective, multicentre non-randomised single-arm study. The primary endpoint was freedom from major adverse cardiac events (MACE) at 30 days post-procedure. Invasive follow-up was scheduled at six months post implantation. A total of 47 patients were enrolled and serial OCT imaging was performed in a subgroup of 18 patients. At the index procedure the lesion success rate was 97.9% (46 patients), the mean acute gain was 1.56±0.43 mm with a mean minimum lumen diameter of 2.48±0.43 mm. Post-implantation OCT imaging revealed a minimal mean prolapse area (0.10±0.06 mm2), mean incomplete stent apposition area (0.12±0.13 mm2) and mean intraluminal mass area (0.05±0.03 mm2). Edge dissections were reported in eight cases (mean dissection width 0.17±0.07 mm proximally and 0.25±0.24 mm distally). At 30-day clinical follow-up, one case of myocardial infarction was reported. At six months, the angiographic mean in-stent late loss was 0.95±0.76 mm. By OCT, a high percentage of struts was covered (97.6 ±15.00 %) with a mean neointimal thickness of 0.31±0.14 mm, all edge dissections were clinically silent and healed. Target lesion revascularisation (TLR) occurred in 11 patients (23.4%) and clinically driven TLR in three of these patients (6.4%). No cases of death or stent thrombosis were reported during the study.
Conclusions: Implantation of the Svelte stent IDS was observed to be safe, feasible and associated with a low acute vascular injury and a high percentage of strut coverage at 6-month follow-up.
Introduction
The novel Svelte stent Integrated Delivery System (IDS; Svelte Medical Systems, New Providence, NJ, USA) is a balloon-expandable, cobalt-chromium, thin-strut stent premounted onto a single lumen fixed-wire delivery system, characterised by a low lesion entry profile (0.012”, 0.305 mm) and crimped stent profile (0.029”, 0.737 mm)1.
The present device represents a new generation of fixed-wire stent technology specifically developed for direct stenting, a technique known to reduce procedural time, radiation dose, contrast administration and costs2-10. In addition, the Svelte stent IDS could provide further savings as it does not require guidewire placement and, with a very low profile1, could be a useful tool in challenging clinical subsets such as tortuous lesions and bifurcation stent-crossings.
Preclinical studies demonstrated similar angiographic results after Svelte stent IDS and Multi-Link Vision Stent (Abbott Vascular, Santa Clara, CA, USA) implantation in the porcine model with comparable late loss and percentage stenosis at 30 and 90 days post-implantation. Consistent with these angiographic data, the vascular response in terms of inflammation and neointimal growth as assessed by histopathology and histomorphology was similar between the two stent types11.
These promising preliminary results have led to the use of this device in humans during the present Svelte Coronary Stent Clinical First-in-Man (FIM) study, with the aim of assessing the safety, feasibility and performance of the Svelte stent IDS for the treatment of de novo coronary lesions. Additionally, the vascular healing process was assessed by high-resolution imaging data provided by OCT12-14.
Methods
STUDY DESIGN AND POPULATION
As described above, the Svelte Coronary Stent Integrated Delivery System (IDS) first-in-man (FIM) trial is a multicentre, prospective, non-randomised, single-arm study to evaluate the safety, feasibility and performance of the Svelte Coronary Stent IDS in the treatment of de novo coronary artery lesions.
The Svelte Coronary Stent IDS has a direct-stenting indication by design. Therefore all lesions were intended to be treated with a direct-stenting strategy, although crossover to a predilatation strategy was permitted. Patients eligible for this study were patients with ischaemic heart disease, meeting all of the general and angiographic inclusion criteria and none of the general or angiographic exclusion criteria (Online Appendix 1). Briefly, patients eligible for enrolment were older than 18 years with stable, unstable angina or a positive functional ischaemia test and a single de novo lesion on a native coronary vessel with a reference vessel diameter ≥2.5 mm and ≤3.5 mm, a target lesion <18 mm and a target lesion stenosis ≥50% and <100%. Patients were entered into the study once the approved study informed consent was signed.
The primary endpoint of the present investigation was freedom from major adverse cardiac events (MACE) at 30 days post-procedure; this was defined as a device-oriented composite (hierarchical order) of cardiac death, myocardial infarction (MI) which was not clearly attributable to a non-target vessel and target lesion revascularisation (TLR).
Secondary endpoints included: 1) a patient-oriented composite (hierarchical order) all-cause mortality, any MI including non-target vessel territory and any repeated revascularisation including all target and non-target vessel; 2) any angiographic restenosis rate and late lumen loss at six months; 3) a composite endpoint of cardiac death and MI at 30 days and six months post-procedure; 4) TLR at 30 and 180 days; 5) rates for each component of the MACE composite endpoint at 30 days and six months post-procedure; 6) any rate of stent thrombosis at six months.
Adjudication of study endpoints was determined by an independent clinical events committee (CEC). The CEC conducted blinded reviews of safety data throughout the study and adjudicated the occurrence of clinical study endpoints, adverse events and reports of device failures in accordance with pre-specified definitions (Online Appendix 2).
In addition, the evaluation of lesion success rate (defined as <30% final residual stenosis of the target lesion by quantitative coronary angiography), procedural time and time to cross the lesion was performed at implant.
An optical coherence tomography (OCT) substudy was per-protocol planned in the Netherlands sites only with OCT imaging performed at post-stent implantation and six-month follow-up. Pre-specified endpoints of the OCT substudy were: 1) stent performance evaluation at post-procedure in terms of minimum lumen area, malapposition, tissue prolapse, dissections (in-stent and at stent-edge), thrombus area; 2) stent performance evaluation at six-month follow-up in terms of minimal lumen area, malapposition, neointimal formation and strut coverage.
STUDY DEVICE
The Svelte stent IDS is a balloon-expandable, cobalt-chromium, thin-strut stent premounted on the Svelte single lumen fixed-wire delivery catheter platform. The lesion entry profile of the shapeable, radiopaque spring tip is 0.012” (0.305 mm) with a crimped stent profile of 0.029” (0.737 mm). Balloon control bands at each end of the balloon control expansion and deflation. The stent IDS delivery catheter has a working length of 145 cm, and includes an integral torqueing device located on the proximal shaft (Figure 1, Figure 2, Figure 3).
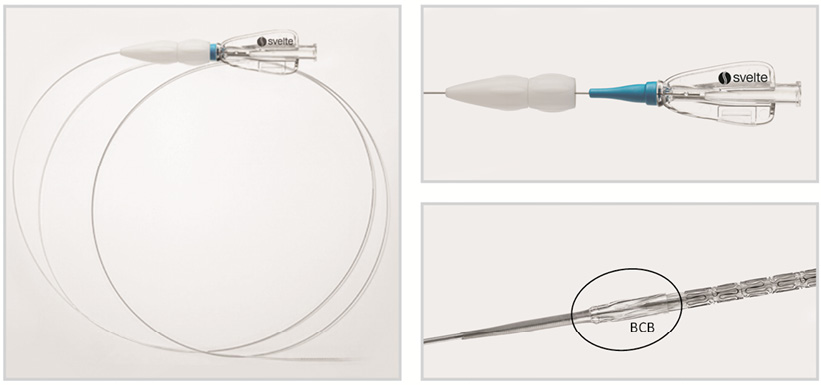
Figure 1. The Svelte stent-on-a-wire integrated delivery system (left panel). Magnifications of the integrated torque system (right upper panel) and of the tip of the stent with balloon control band (BCB) (right lower panel).

Figure 2. Svelte balloon control bands. Balloon control bands at the proximal (right panel) and distal (left panel) end of the balloon were designed to control balloon expansion and deflation during stent deployment in order to reduce the vascular injury at the stent edges.

Figure 3. The Svelte stent integrated delivery system has a very low profile in comparison with other bare metal stents (ML Vision and Driver stent).
QCA ANALYSIS
Quantitative coronary angiography (QCA) analyses were performed with the Coronary Angiography Analysis System (Pie Medical Imaging, Maastricht, Netherlands). The following QCA parameters were computed: pre-procedural reference vessel diameter (RVD) calculated with interpolated method, minimum luminal diameter (MLD), percentage diameter stenosis (%DS), in-stent acute gain and binary restenosis. Late loss was defined as the difference between the post-procedural and follow-up minimal luminal diameter.
OCT ACQUISITION AND ANALYSIS
Fourier-domain OCT (C7XR system; St. Jude/LightLab Imaging Inc., Westford, MA, USA) and time-domain OCT (M3 system; St. Jude/LightLab Imaging Inc.) systems were used in this study. Vessels were imaged with an automated pullback system at 20 mm/sec (C7XR system) and 3.0 mm/sec (M3 system). Cross-sectional images were acquired at 100 frames/sec for C7 and at 20 frames/sec for M3. Imaging acquisitions were evaluated by an independent core laboratory (Cardialysis, Rotterdam, The Netherlands) and measurements were performed off-line by two independent observers using LightL ab imaging offline software. Cross-sections at 1 mm intervals within the stented segment were analysed; in each cross-section, lumen contours were drawn with a semi-automated detection algorithm and additional manual corrections were performed if needed. The stent contours were traced using a multiple point detection function, struts were considered uncovered if any part of the strut was exposed to the lumen and incomplete stent apposition (ISA) was defined as separation of at least one strut from the vessel wall. To assess ISA, the distance from the endoluminal middle point of the strut to the vessel wall was assessed. If the distance was observed to be greater than the strut thickness (strut thickness 81 μm), this was considered as incompletely apposed. Neointimal hyperplasia (NIH) area at follow-up was calculated as stent area minus lumen area. Tissue prolapse was defined as protrusion of tissue between adjacent struts, with a distance from the arc connecting the struts and the greater extent of protrusion >50 μm. Intra-stent dissection was defined as a disruption of the luminal vessel surface in the stent segment. It can appear in two forms: (a) dissection, the vessel surface is disrupted and a dissection flap is visible; (b) cavity, the vessel surface is disrupted and an empty cavity can be seen. For the purpose of simplification, they will both appear in the present study as in-stent dissection; the flap length and the length of region containing dissection are reported. Edge dissection was defined as disruption of the luminal vessel surface within the 5 mm proximal and distal to the stent. For edge dissection analysis, dissection width15,16 and both the flap length, measured from the joint point with the vessel wall to the tip of the flap, and the length of region containing dissection were assessed (Figure 4).
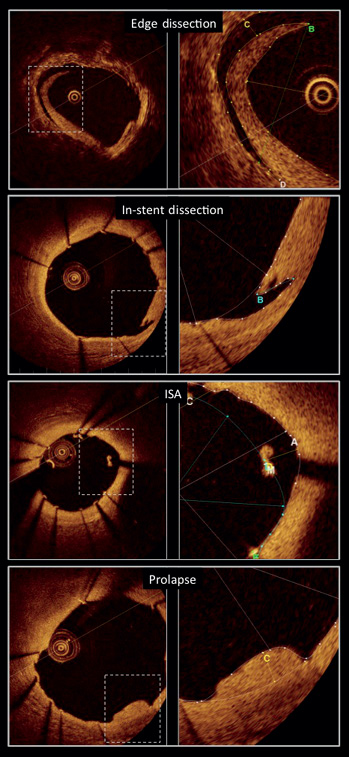
Figure 4. OCT quantitative measurements of edge dissection, in-stent dissection, incomplete stent apposition (ISA) and plaque prolapse.
A dedicated score of neointimal healing (“healing score”) combining three components (intraluminal masses, malapposition and strut coverage) was applied for analysis. This “healing score” is a weighted index that was structured as follows:
1. Presence of filling defect (%ILD) is assigned weight of “4”.
2. Presence of both malapposed and uncovered struts (%MN) is assigned a weight of “3”.
3. Presence of uncovered struts alone (%N) is assigned a weight of “2”.
4. Presence of malapposition alone (%M) is assigned a weight of “1”.
Neointimal healing score=(%ILD*‘4’)+(%MN*‘3’)+(%N*‘2’)+(%M*‘1’).
In addition, to provide further insights on plaque characteristics, two experienced operators assessed in agreement the plaque located behind the stent at each millimetre of the OCT pullback. Four plaque types were defined as follows:
Fibrous plaque, if the plaque was predominantly composed of fibrous tissue (high backscattering and homogeneous signal) and no relevant lipid pool or calcifications were observed.
Fibro-calcified plaque, if the presence of calcifications (signal-poor or heterogeneous region with a sharply delineated border) was observed.
Lipid plaque, if lipid content (signal-poor region within an atherosclerotic plaque, with poorly delineated borders) was detected in an area superior to one quadrant of the frame (90 degrees).
Mixed lipid and calcified plaque, defined as a lipid plaque containing calcifications.
The reproducibility of the two observers was assessed in 80 frames that were analysed twice by the two observers at one-week intervals.
Statistical analysis
Continuous variables are presented as mean±standard deviation, categorical variables as count and percentage. Paired comparisons of continuous variables within groups between different time points were performed with Wilcoxon’s signed rank test. A two-sided p-value ≤0.05 was considered statistically significant. All statistical tests were performed with SPSS, version 15.0 for Windows (SPSS; IBM Corp., Armonk, NY, USA). The intra-observer agreement for qualitative measurements was evaluated using Cohen’s Kappa test for concordance. A kappa value <0 was defined as poor agreement, 0-0.20 slight agreement, 0.21-0.40 fair agreement, 0.41-0.60 moderate agreement, 0.60-80 good agreement, 0.81-1.0 excellent agreement.
Results
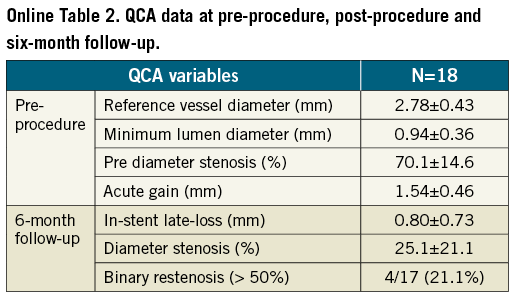
A total of 47 patients were enrolled in the present study. Baseline clinical characteristics are reported in Table 1. The overall lesion success rate was 97.9% (obtained in 46 patients). The mean procedural time was 36.7±22.40 minutes, with a mean time to cross the lesion of 3.50±3.44 minutes and a mean fluoroscopy time of 15.2±14.39 minutes. At post-procedure, the mean acute gain was 1.56±0.43 mm with a mean minimum lumen diameter of 2.48±0.43 mm.
At six-month angiographic follow-up the mean in-stent late loss was 0.95±0.76 mm. Binary restenosis was observed in 11 cases with an angiographic restenosis rate of 23.4% (Table 2).
The primary endpoint of freedom from major adverse cardiac events at 30 days post-procedure was reached in 97.9% of the patients, with a reported case of myocardial infarction within 30 days post-procedure (Table 3). This event occurred in a patient who was treated for a non-target vessel lesion within 30 days. In the same session, following a positive FFR (FFR= 0.76), a balloon dilatation was performed in the target lesion with a drug-eluting balloon; this was associated with a distal dissection of the vessel provoked by the tip of the wire. The patients experienced enzyme elevation. The event was adjudicated as non-Q-wave MI.
From 30 days to the six-month follow-up no changes were observed in the composite endpoint of cardiac death and MI (2.1%). A total of 11 patients underwent target lesion revascularisation; clinically-driven TLR occurred in three of those patients (6.4%) (Figure 5). No cases of stent thrombosis were reported during the study (Table 3).
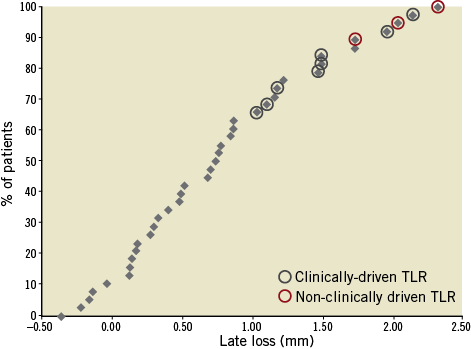
Figure 5. Cumulative frequency distribution curve of late loss. Patients who underwent clinically-driven and non-clinically driven TLR are highlighted.
A total of 18 patients underwent OCT imaging at post-stent implantation and six-month follow-up (baseline clinical characteristics and QCA data at pre-procedure and six-month follow-up are presented in Online Table 1 and Online Table 2). OCT parameters reflecting the acute effects of stent implantation on the vessel wall or acute stent performance such as post-implantation mean plaque prolapse area (0.10±0.06 mm2), mean incomplete stent apposition area (0.12±0.13 mm2), mean intraluminal mass area (0.05±0.03 mm2) and mean length of in-stent dissection (0.37±0.19 mm) were minimal and no longer observed at follow-up (Table 4). Eight cases of post-implantation edge dissection were reported, with a mean dissection flap of 1.05±0.64 mm proximally to the stent and 0.68±0.45 mm distally, the mean dissection width was 0.17±0.07 mm proximally and 0.25±0.24 mm distally. Of note seven out of the eight cases with edge dissection were cases in which a high-pressure post-dilatation was performed after the stent placement. All edge dissections were clinically silent and healed after six months (Figure 6).
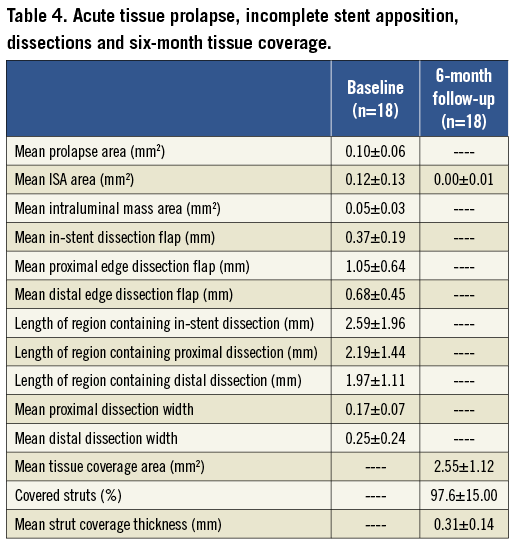
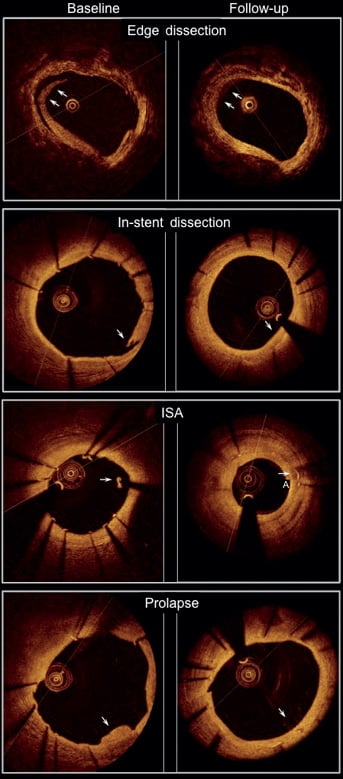
Figure 6. OCT images showing edge dissection, in-stent dissection, incomplete stent apposition (ISA) and plaque prolapse (arrows) at post-implantation (left panel) and at six-month follow-up (right panel). Parameters reflecting acute effects of stent implantation on the vessel wall such as post-implantation mean plaque prolapse area, mean incomplete stent apposition (ISA) area and the mean length of in-stent dissection flap were minimal and no longer observed at follow-up.
The analysis of the plaque located behind the stent was performed in a total of 333 frames; 82 fibrous plaques, 136 lipid plaques, 26 calcified plaques and 89 mixed plaques were detected. In all patients at least six consecutive millimetres of lipid or mixed lipid-calcified plaques were observed. Six out of eight patients who developed edge dissections showed lipid or mixed plaques at the site of the dissection. A good agreement was noted between the estimations of the two observers with regard to the composition of the plaque in the 80 frames that were analysed twice (kappa statistic: 0.79).
At six-month follow-up, a high percentage of struts appeared covered (97.6 ±15.0%) (Figure 7), the mean thickness of strut coverage was 0.31±0.14 mm (Table 4).
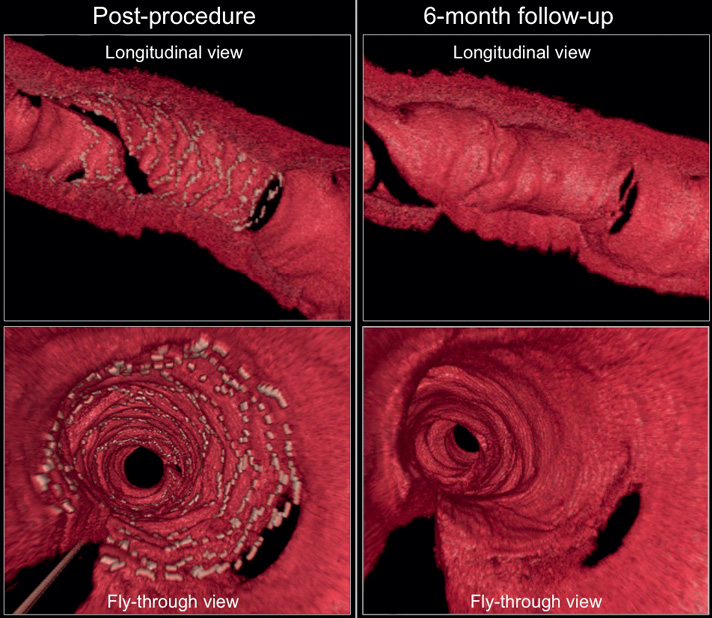
Figure 7. Serial three-dimensional OCT images at post-procedure (left panel) and six-month follow-up (right panel). The novel Svelte stent is completely covered at six-month follow-up, with the struts at the side branch ostium also appearing to be well covered (upper right panel) and without excessive neointimal proliferation (lower right panel).
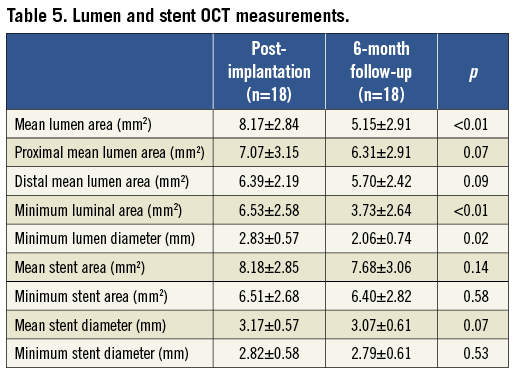
No changes were observed in mean stent area (8.18±2.85 mm2 vs. 7.68±3.06 mm2, p=0.14), minimum stent area (6.51±2.68 mm2 vs. 6.40±2.82 mm2, p=0.58) and mean stent diameter (3.17±0.57 mm vs. 3.07±0.61 mm, p=0.07) (Table 5). Neointimal growth led to a reduction in mean lumen area (8.17±2.84 mm2 vs. 5.15±2.91 mm2, p<0.01), minimum luminal area (6.53±2.58 mm2 vs. 3.73±2.64 mm2, p<0.01) and minimum lumen diameter (2.83±0.57 mm vs. 2.06±0.74 mm, p=0.02) over time (Table 5). The healing score in the present population at post-stent implantation was observed to be 180.2±61.9; at six-month follow-up it was found to be reduced to 5.8±10.2 (p<0.01).
Discussion
The present report is the first evaluation of the Svelte coronary stent IDS after implantation in humans. This novel fixed-wire device intended for direct stenting was designed to allow reduction in procedural time, fluoroscopy time and contrast usage that are clinically relevant issues for selected high-risk patients17-20. Beside these theoretical advantages, this stent technology shows a very low profile that could be additionally beneficial for the treatment of challenging lesions (i.e., bifurcations, tortuous vessels, distal lesions, long lesions, etc.) often observed especially in the above-mentioned high-risk patients.
In the present study an optimal lesion success rate (97.9%) with an overall short procedural time, fluoroscopy time and time to cross the lesion was observed. Even though the single arm design of the present investigation does not allow a direct comparison with other devices, these data suggest a good deliverability of the Svelte stent. Patients reached the pre-specified primary endpoint of freedom from major adverse cardiac events at 30 days post-procedure in 97.9% of the cases. This observation, in addition to the encouraging procedural data, demonstrates safety and feasibility of the Svelte coronary stent IDS.
The first-in-man implantation follows a preclinical evaluation of the Svelte stent in porcine coronary arteries. In the healthy animal model, vascular healing and a tissue response appeared substantially equivalent to those observed after implantation of another standard bare metal stent (ML Vision) in the same setting, with comparable luminal losses at 30 and 90 days21. However, in the present investigation, the late lumen loss reported at six-month follow-up after implantation in humans with coronary artery disease appeared to be slightly higher compared to historical data after implantation of other bare metal stents22,23. Notably, this increased late loss did not translate into a higher number of clinically-driven target lesion revascularisations (6.4%), which would be comparable with other series investigating newer-generation, thin-strut, bare metal stents24,25. This effect could be partially explained by the fact that, in the present study, only stents with a diameter of 3.0 mm were used and deployed in large vessels, thus reducing the clinical impact of the neointimal growth.
The present report is also the first attempt to provide information on the healing process after Svelte stent implantation using OCT, a high-resolution intravascular imaging modality that allows a detailed assessment of the anatomical relations between stent and vessel wall26, and which has been previously used to detect signs of vascular injury after stent deployment27.
OCT data analysis showed low vascular injury following the Svelte stent IDS placement, expressed by a minimal amount of plaque prolapse and in-stent dissection observed at the index procedure. Of note, tissue prolapse area was considerably inferior compared to previous historical OCT observations in BMS27,28, and both plaque prolapse and in-stent dissection were no longer detectable at six-month follow-up.
While plaque prolapse is commonly considered a sign of vascular injury, it has not been associated with an increase in events28. Porto at al recently demonstrated that acute in-stent dissection is an independent predictor of periprocedural myocardial infarction (MI)29. These results may be related to the fact that endothelial disruption and intimal flap could act as a trigger for platelet aggregation. The same group also showed that post-procedural intraluminal masses are an independent predictor of type VIa MI, presumably in association with distal embolisation of thrombotic material29. According to this background –and considering that the in-stent dissections and, especially, the amount of intraluminal masses after Svelte stent IDS placement were minimal as well as being in line with or inferior to previous reports investigating BMS acute performance27– our results may suggest a safety profile of the Svelte stent IDS similar to other BMS, in terms of acute vascular injury.
The safety profile of the Svelte stent is also confirmed by an important reduction in the value of the healing score from baseline to follow-up. Of note, this is the first time that this healing score has been applied to a population treated with bare metal stents. The healing score has been previously evaluated in stable patients treated with everolimus-eluting biocompatible durable polymer and everolimus-eluting bioresorbable vascular scaffolds (BVS) with a healing score of 10.8±15.3 and 9.0±9.4, respectively, at six-month follow-up (data on file; Cardialysis, Rotterdam, The Netherlands).
However, these data, due to the small number of patients included in the presents study, need to be confirmed by larger investigations.
An additional specific technical characteristic of the novel Svelte stent IDS is the presence of control bands at the two extremities of the stent balloon (Figure 2) designed to reduce vascular injury at the stent edges during balloon inflation and stent deployment1. Outward balloon expansion is thought to be a possible cause of vascular damage, perhaps inducing endothelial disruption and edge dissection. Furthermore, previous pathological investigations proposed plaque disruption in the proximity of the stented arterial segment as a mechanism of stent thrombosis, especially in the presence of necrotic core-rich plaques30,31. In the present report, out of the eight cases with edge dissection, seven cases were reported in patients who underwent post-dilation after Svelte stent IDS deployment. Still, in all other cases without post-dilatation, an edge dissection was only detected once by OCT. According to these observations in this population, a cause-effect association for post-dilatation (associated with high-risk plaque at the stent edge in six cases) and edge dissection could not be excluded. Of note, all edge dissections were non-flow-limiting, angiographically not visible, clinically silent and completely healed at follow-up.
At six-month follow-up a high percentage of stent struts were covered, no significant changes in stent area and stent diameters were observed but a significant reduction in mean lumen area, mean and minimum lumen diameter was seen, associated with neointimal tissue growth. The mean thickness of that neointimal tissue was observed to be 0.31±0.14 mm. Chen et al compared neointimal growth after BMS and sirolimus- eluting stent (SES) implantation using OCT, reporting a neointimal thickness comprised between 0.20 mm and 0.59 mm for BMS32; similar results were also reported by Goto et al, with a mean neointimal growth of 0.35±0.17 mm observed in patients imaged with OCT at five to 10 months post-BMS implantation33. These observations show a substantial consistency between our data on neointimal formation after Svelte stent IDS placement and previous data with other bare metal stents.
However, it should be highlighted that if the value of 0.31±0.14 mm mean neointimal thickness as derived by OCT is in line with the angiographic late loss (0.80±0.73 mm) also observed in the OCT subpopulation (Online Table 2), the entire population enrolled in the study showed a slightly higher overall late loss (0.95±0.76 mm).
In conclusion, data observed in the present study suggest the safety and feasibility of the implantation of the thin-strut Svelte stent IDS with promising results in relation to acute vascular injury. Still, at the current status of this technology, the reduction of the neointimal proliferation appears to be the most important target for potential improvement of the device. A novel sirolimus-eluting Svelte coronary stent IDS is currently under evaluation and in the near future data on the implantation in humans will be available. This novel drug-eluting version of the current device may aid in further understanding the real potential of the fixed wire technology in the clinical scenario.
Limitations
The Svelte FIM study evaluated the feasibility and safety of the Svelte stent implantation in humans and no direct comparison with other bare metal stents was performed.
The theoretical advantages of this new technology, such as reduction in procedural time, fluoroscopy time, contrast use and costs, were not per protocol planned as the primary aim of the current study. Plaque characterisation was performed by OCT only after stent placement. The present study was conducted on a small number of patients and results should be considered exploratory and hypothesis-generating.
Conclusion
Implantation of the Svelte stent IDS for treatment of de novo coronary artery lesions was observed to be safe and feasible, with low vascular injury after deployment and a high percentage of strut coverage at six-month follow-up. Further studies with larger populations and longer follow-up are needed to fully evaluate the performance of this novel device.
Conflict of interest statement
R. Spencer, J. De Schepper and M. Pomeranz are employees of Svelte Medical. The other authors have no conflicts of interest to declare. The Guest Editor, Antonio Colombo, has no conflict of interest to declare.
Online appendix 1
INCLUSION CRITERIA
GENERAL INCLUSION CRITERIA
Patients included in the present study were male or female patients older than 18 years, presenting with stable or unstable angina pectoris or a positive functional ischaemia study, with an understanding of the nature of the procedure and providing written informed consent prior to the catheterisation procedure.
ANGIOGRAPHIC INCLUSION CRITERIA
Patient indicated for elective stenting of a single stenotic lesion in a native coronary artery with a reference vessel diameter ≥2.5 mm and ≤3.5 mm, a lesion length < 18 mm and a percentage stenosis ≥50% and <100% in by visual estimation.
EXCLUSION CRITERIA
GENERAL EXCLUSION CRITERIA
Exclusion criteria comprised patient enrolment in another investigational device or drug trial that has not completed the primary endpoint or that clinically interferes with the current study endpoints. Any previous coronary interventional procedure within 30 days prior to the procedure. Patients requiring a staged procedure of the target vessel within six months, or a staged procedure of a non-target vessel within 30 days post-procedure. Target lesions requiring treatment with a device other than PTCA prior to stent placement (such as, but not limited to, directional coronary atherectomy, rotational atherectomy). Previous bare metal stent (BMS) deployment anywhere in the target vessel within the past six months. Any DES deployment in the target vessel within the previous nine months. Any previous stent placement within 10 mm (proximal or distal) of the target lesion. Comorbid condition(s) that could limit the patient’s ability to participate in the trial or to comply with follow-up requirements, or impact the scientific integrity of the trial. Concurrent medical condition with a life expectancy of less than 12 months. Documented left ventricular ejection fraction (LVEF) <30% at the most recent evaluation. Evidence of an acute myocardial infarction (MI) within 72 hours of the intended trial procedure. History of cerebrovascular accident or transient ischaemic attack in the last six months. Leukopenia (leukocytes <3.5×109/L). Neutropenia (absolute neutrophil count <1000/mm3) ≤3 days prior to enrolment. Thrombocytopenia (platelets <100,000/mm3) preprocedure. Active peptic ulcer or active gastrointestinal (GI) bleeding. History of bleeding diathesis or coagulopathy or inability to accept blood transfusions. Known hypersensitivity or contraindication to aspirin, heparin or bivalirudin, clopidogrel or ticlopidine, cobalt, nickel, L-605 cobalt chromium alloy or sensitivity to contrast media, which cannot be adequately pre-medicated. Serum creatinine level >2.5 mg/dL within seven days prior to index procedure.
ANGIOGRAPHIC EXCLUSION CRITERIA
Angiographic exclusion criteria were unprotected left main coronary artery disease. Target vessel exhibiting multiple lesions with greater than 40% diameter stenosis outside of a range of 5 mm proximal and distal to the target lesion based on visual estimate or on-line quantitative coronary angiography (QCA). Target lesion exhibiting an intraluminal thrombus. Aorto-ostial lesions. Target lesion with side branches >2.0 mm in diameter. Severely calcified target lesion. Target lesions judged to have a lesion that prevents complete inflation of an angioplasty balloon or proper placement of the stent or stent delivery system.
Online Appendix 2
DEFINITIONS
Cardiac death is defined as any death due to proximate cardiac cause (e.g., MI, low-output failure, fatal arrhythmia), unwitnessed death and death of unknown cause, and all procedure-related deaths, including those related to concomitant treatment, will be classified as cardiac death. All deaths are considered cardiac unless an unequivocal non-cardiac cause can be established. Specifically, any unexpected death, even in patients with coexisting potentially fatal non-cardiac disease (e.g., cancer, infection), should be classified as cardiac.
Major adverse cardiac events (MACE) are defined as cardiac death, myocardial infarction (MI) (Q wave and non-Q wave), emergent bypass surgery, or clinically driven target lesion revascularisation (TLR) by percutaneous or surgical methods.
Q wave MI (QWMI) is defined according to one of the following criteria:
1) Chest pain or other acute symptoms consistent with myocardial ischaemia and new pathological Q waves in two or more contiguous ECG leads as determined by an ECG core laboratory or independent review of the CEC, in the absence of timely cardiac enzyme data.
2) New pathologic Q waves in two or more contiguous ECG leads as determined by an ECG core laboratory or independent review of the CEC and elevation of cardiac enzymes above normal. In the absence of ECG data, the CEC may adjudicate Q wave MI based on the clinical scenario and appropriate cardiac enzyme data.
Non-Q Wave MI (NQWMI) is defined as elevated CK ≥ 2X the laboratory upper limit of normal with the presence of an elevated CK-MB (any amount above the laboratory upper limit of normal) in the absence of new pathological Q waves. If CK and CK-MB not available, elevation of ctn ≥ 3 times the laboratory upper limit of normal will be considered a non-Q wave MI.
Stent thrombosis will be reported according to the ARC definitions.
Target lesion revascularisation (TLR) is defined as any repeat percutaneous intervention of the target lesion or bypass surgery of the target vessel performed for restenosis or other complication of the target lesion. All TLRs should be classified prospectively as clinically indicated or not clinically indicated by the investigator prior to repeat angiography. An independent angiographic core laboratory should verify that the severity of percent diameter stenosis meets requirements for clinical indication and will overrule in cases where investigator reports are not in agreement. The target lesion is defined as the treated segment from 5 mm proximal to the stent and to 5 mm distal to the stent. Clinically-driven target lesion revascularisation (TLR) is defined as revascularisation at the target site associated with positive functional ischaemia study or ischaemic symptoms and an angiographic minimal lumen diameter stenosis ≥50% by QCA, or revascularisation of a target site with diameter stenosis ≥70% by QCA without either angina or a positive functional study.
