Abstract
Aims: Restenosis is a frequent complication of coronary stent implantation, especially bare metal stent (BMS) implantation. The everolimus-eluting stent (EES) has previously been shown to be efficacious in the treatment of de novo lesions. We performed this study to evaluate clinical, angiographic and IVUS results after EES implantation for the treatment of BMS ISR.
Methods and results: XERES was a prospective, multicentre, nationwide study, enrolling 97 consecutive patients with in-stent restenosis (ISR) after BMS implantation across 20 centres in France. Suitable lesions had a reference vessel diameter between 2.5 mm and 4 mm, a length ≤22 mm and a diameter stenosis between 50 and 100%. The primary endpoint was angiographic in-stent late loss (LL) as determined by quantitative coronary angiography (QCA) at nine-month follow-up. QCA was required to be performed in each included patient and IVUS was performed in a subgroup of 27 patients. At nine-month follow-up, the in-stent late loss was 0.35±0.63 mm. The rate of in-stent binary restenosis was 12.22%, including two complete occlusions. The average volume of neointimal hyperplasia was 15.6±9.9 mm3. The in-stent percent volume obstruction was 8.5±5.2%. The in-segment percent area and diameter obstruction were 32±17% and 27±11%, respectively. Two initial malappositions were persistent and two other patients had late acquired stent malapposition. The cumulative incidence of major adverse cardiac events (MACE) was 10.1%.
Conclusions: EES for the treatment of bare metal in-stent restenosis seemed safe and efficacious.
Introduction
Restenosis is the most common complication associated with coronary stent implantation1. Some studies that have evaluated the local application of antiproliferative drugs (sirolimus and paclitaxel) for the prevention of restenosis via a stent delivery system have shown that these therapies successfully inhibit the development of neointimal hyperplasia and reduce restenosis and associated clinical events2,3.
Everolimus is an effective antiproliferative agent that inhibits growth factor-stimulated cell proliferation by causing cell cycle arrest in the last G1 stage in the cell cycle4. The everolimus-eluting stent (EES) is a second-generation drug-eluting stent which had been shown to be very effective for the treatment of de novo coronary lesions through many studies, including FUTURE (First Use To Underscore REduction in restenosis with everolimus) I and II5-8, and, more recently, the SPIRIT (safety and performance of an everolimus-eluting coronary stent versus a paclitaxel-eluting coronary stent in patients with de novo native coronary artery lesions) family9-13 and COMPARE (comparison of the second-generation everolimus-eluting and paclitaxel-eluting stents in real-life practice)14 studies. In the large-scale, prospective randomised trial, SPIRIT III, an everolimus-eluting stent compared with a paclitaxel-eluting stent resulted in reduced angiographic late loss, a non-inferior rate of target vessel failure, and fewer major adverse cardiac events during one year of follow-up12.
However, the efficacy and safety of the second-generation EES for the treatment of BMS (bare metal stent) ISR (in-stent restenosis) lesions remains less well defined15. The present study, XERES, will evaluate clinical, angiographic (QCA) and intravascular ultrasound (IVUS) results after EES implantation for BMS ISR. We present here the nine-month results of this trial.
Methods
STUDY DESIGN
This study was designed to evaluate the angiographic results at nine months after XIENCE stent implantation for BMS ISR. It is a prospective observational cohort study with clinical and angiographic follow-up. A subgroup of 27 patients was also subjected to IVUS investigation. This study was approved by the Toulouse CPP (Comité de Protection des Personnes) (reference 1-08-25 of 09/10/08).
PATIENT POPULATION
From April 2009 to July 2010, in 20 centres in France, this prospective, multicentre, nationwide study enrolled 102 consecutive patients with in-stent restenosis (ISR) in their previous BMS implant.
Patients were eligible for enrolment if they had a first and unique in-stent restenosis (focal, diffuse or proliferative) in a bare metal stent with or without other lesions on any coronary vessel. The target lesion reference vessel diameter was required to be between 2.5 mm and 4 mm by visual estimation, ≤22 mm in length, have a visually estimated stenosis between 50 and 100% of the luminal diameter, and a Thrombolysis In Myocardial Infarction (TIMI) flow of 1 or more. Patients were excluded if they had a known diagnosis of acute myocardial infarction three days prior to baseline procedure, a left ventricular ejection fraction of less than 30%, were awaiting a heart transplantation or had a known hypersensitivity or contraindication to aspirin, heparin, bivalirudin, clopidogrel, ticlopidine, different components of a stent (cobalt, chromium, nickel, tungsten, everolimus, acrylic and fluoro polymers) or contrast sensitivity that could not be adequately pre-medicated. Additionally, patients with ISR within an aorto-ostial, left main or coronary bypass location, or at a bifurcation including a side branch with a diameter >1.5 mm, heavy calcification, total occlusion of the target vessel, or an angiographically detectable thrombus in the target vessel were not eligible. Patients with previous failure for multiple lesions treated by angioplasty and patients with a restenosis lesion previously treated by another device (except by a balloon catheter), including cutting balloon, an atherectomy device, a laser, brachytherapy or other eluting stent were not eligible. All included patients gave written informed consent.
FOLLOW-UP
Patients were required to be reviewed at 1, 8, 9, 12 months and 2 years following the procedure. Three angiographic records (QCA) were planned for all patients at pre-implantation, post-implantation and at nine-month follow-up, and IVUS records in a subset of 27 patients (of three specialised cardiologic units) were performed after implantation and at nine months. This report focuses on the nine-month follow-up results.
THE EVEROLIMUS-ELUTING STENT
XIENCE V® (Abbott Vascular, Santa Clara, CA, USA) and XIENCE PRIME™ (Abbott Vascular) stents are composed of four elements: the platform (L605 cobalt-chromium alloy metal stent of MULTI-LINK 8 range); the non-erodible polymeric matrix covering the platform; the drug, everolimus, an antiproliferative molecule associated with the polymer matrix; and the stent delivery catheter with balloon dilatation.
XIENCE PRIME differs from its predecessor XIENCE V in: the design of the mesh of the metal platform; the installation system which has a higher balloon burst pressure; the catheter body with shorter shoulders, a wider hypotube and an hypotube junction in stainless steel; and a range of larger sizes (with new lengths of 33 and 38 mm).
The polymer matrix and the total concentration of the active drug (100 μg/cm²) are similar in both XIENCE V and XIENCE PRIME, and the majority of the drug is released within the first 28 days.
STUDY ENDPOINTS
The primary endpoint was angiographic in-stent LL as determined by quantitative coronary angiography at nine-month follow-up.
Secondary endpoints included in-stent and in-segment percentage volume obstruction (%VO) (IVUS), in-stent neointimal volume (IVUS), in-stent and in-segment binary restenosis (QCA), in-segment LL (QCA), rate of late-acquired incomplete apposition of the stent (IVUS) at nine-month follow-up; number of stent thromboses, target lesion revascularisation (TLR), target vessel revascularisation (TVR) and major adverse cardiac events (MACE: cardiac death, acute coronary syndrome, stent thrombosis, repeat angioplasty, bypass surgery, stroke) rate at 1, 8, 9, 12 and 24 months follow-up.
All deaths that could not be clearly attributed to another cause were considered cardiac deaths. Myocardial infarction was defined according to the World Health Organization definition based on creatine kinase and creatine kinase-MB rise. Stent thrombosis was defined as angiographic confirmation of the presence of thrombus originating in the stent or in the segment 5 mm proximal or distal to the stent and the presence of at least one of the following criteria within a 48-hour follow-up period: 1) acute onset with ischaemic symptoms at rest; 2) new ischaemic ECG changes suggestive of acute ischaemia; 3) a typical rise and fall in cardiac biomarkers; 4) non-occlusive thrombus; or 5) occlusive thrombus. Incomplete stent expansion was defined as: 1) in-stent minimal lumen area <80% of the average reference lumen area or <90% of lumen area of the reference segment with the lowest lumen area; 2) in-stent lumen area of proximal stent entrance <90% of proximal reference lumen area. All MACE were adjudicated by the clinical events committee.
QUANTITATIVE CORONARY ANGIOGRAPHY EVALUATION
The QCA was performed using the QCA-CMS™ quantification system, version 4.0 (Medis medical imaging systems bv, Leiden, The Netherlands). In each patient, in-stent and in-segment areas were analysed. In-stent was defined as within the margins of the stent, while in-segment was defined as located within the margins of the stent and 5 mm proximal or distal to the stent. The following QCA parameters were computed: TIMI flow, minimal luminal diameter (MLD), reference vessel diameter (RVD), percentage diameter stenosis (DS), length, and Mehran classification of in-stent restenosis16. Reading was centralised. Binary restenosis (BR) was defined in every segment as diameter stenosis ≥50% at follow-up. Late loss (LL) was defined as the difference between MLD post-procedure and MLD at follow-up. Acute gain was defined as the difference between MLD post-procedure and MLD pre-procedure.
INTRAVASCULAR ULTRASOUND ANALYSIS
IVUS examination was performed in a subgroup of 27 patients. IVUS images were acquired in the usual manner of intravascular ultrasound (probe 45 MHz). All IVUS measurements were performed using the EchoPlaq v3.0.53 analysis system (INDEC Systems, Inc., Santa Clara, CA, USA). After deployment of the prosthesis and injection of 0.5 mg of intravenous nitroglycerine, the IVUS catheter was placed in the downstream of the coronary vessel and image recording was performed during withdrawal of the catheter at a constant speed of 0.5 mm/s (motor system). At the reference segments, surfaces, luminal and vessel diameters were measured at five levels (1 mm apart). The volumes were calculated using the Simpson rule. Reading was centralised. In each patient, in-stent and in-segment area were analysed. The in-stent neointimal volume was calculated as the difference between stent volume (SV) and lumen volume (LV). The percentage obstruction of the stent volume was calculated as [(in-stent neointimal volume)/(stent volume)]*100. Late-acquired incomplete apposition was defined as incomplete apposition (one or more stent struts separated from the vessel wall with evidence of blood speckles behind the strut on ultrasound) of the stent at follow-up which was not present post-procedure.
DATA MANAGEMENT
Independent study monitors verified 100% of case report form data on-site. All clinical events (death, myocardial infarction, TLR, TVR) were collected and adjudicated by an independent clinical events committee after review of original source documentation.
STATISTICAL METHODS
All analyses were performed on the intent-to-treat (ITT) population which was defined as the population of patients treated with XIENCE stents and having at least one post baseline data (follow-up data). Missing data were not replaced and were treated as such by the analyses used.
The sample size for the study was determined based on the primary endpoint of in-stent LL at nine-month follow-up. A population of 96 patients allows us to estimate an in-stent late loss mean, with an error margin of 0.05 at 95% confidence. With the assumption that some data will be unusable, 10% more patients were included. So, in this study, 102 patients were included. A significance level of 5% was fixed for the entire study. Comparisons were bilateral. Descriptive statistics included, for quantitative variables, size, average, standard deviation, extreme values, median and quartiles if relevant, and, for categorical variables, numbers and percentages of the different classes. Subgroup comparisons were performed using parametric tests (Student’s t-tests and chi-square tests) or non-parametric tests (Wilcoxon two-sample or Fisher’s exact tests) when the validity conditions of parametric tests were not met.
The primary endpoint analysis and the secondary endpoints measured at nine months were planned at nine-month follow-up. The other secondary endpoints (measured at one month, eight months, nine months and 12 months) were planned to be analysed if at least 75% of expected data were available.
Results
PATIENT CHARACTERISTICS
Between April 2009 and July 2010, 102 patients with in-stent restenosis (ISR) after a BMS implant were enrolled in 20 French sites. Five patients were excluded due to major protocol deviations (four patients with more than one restenosis lesion and one patient with aorto-ostial lesion). However, two patients with two focal and contiguous ISR lesions were included in the study after acceptance by the independent clinical events committee. Nine-month follow-up was obtained in 95 patients with one withdrawal and one cardiac death between eight and nine months.
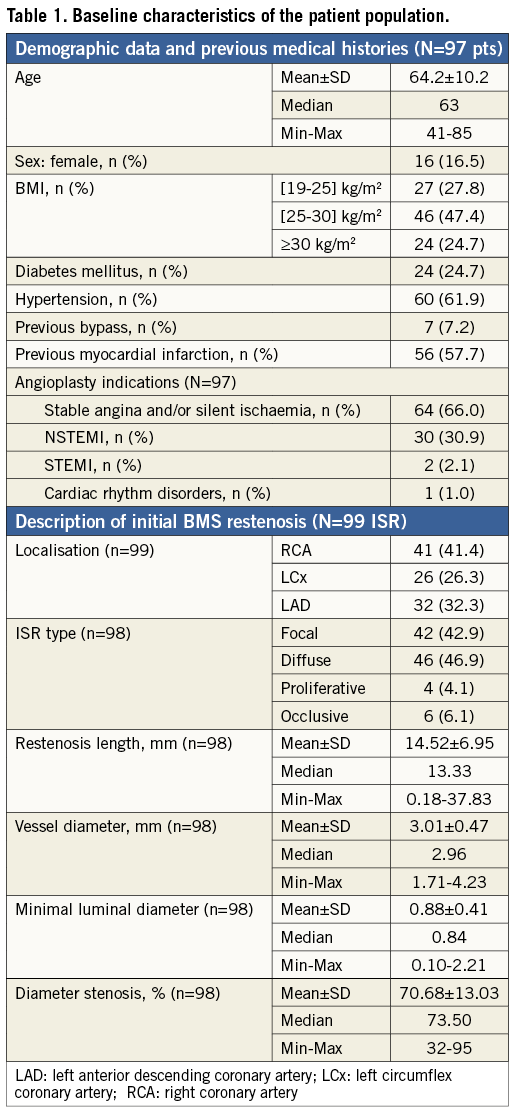
The baseline demographic data, previous medical histories and BMS restenosis characteristics are summarised in Table 1.
PROCEDURAL CHARACTERISTICS
The procedure was elective for stable angina or silent ischaemia in 52 patients. ISR type was focal in 43%, diffuse in 47%, proliferative in 4% and occlusive in 6%.
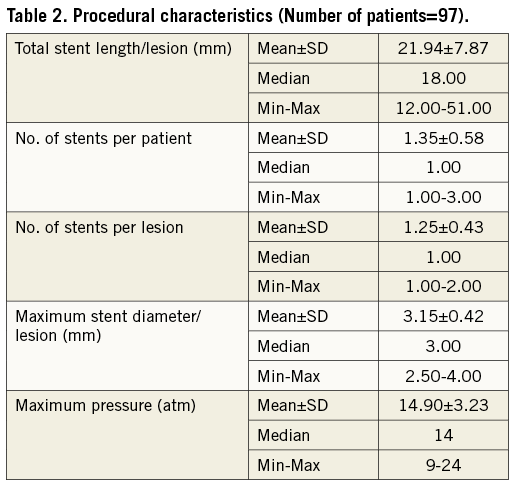
Ninety-nine lesions of BMS restenosis were treated with a total of 113 XIENCE stents (112 XIENCE V and 1 XIENCE PRIME). Procedural success was 98% (97/99 lesions). As shown in Table 2, 1.35±0.58 study stents were implanted per patient and mean stent deployment pressure was 14.90±3.23 atm. All the patients were treated with dual antiplatelet therapy for a minimum of six-month follow-up; at nine-month outcome, 100% of patients were treated with at least one antiplatelet agent (acetylsalicylic acid in the majority or clopidogrel or ticlopidine).
QUANTITATIVE CORONARY ANGIOGRAPHY ANALYSIS
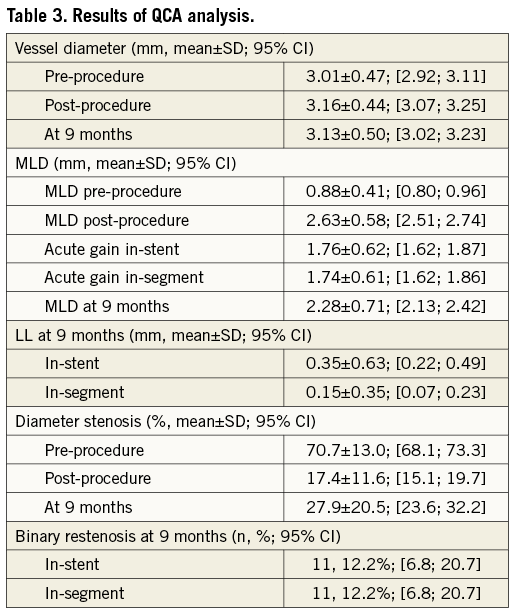
Angiographic control at nine-month follow-up was completed in 86.3% (88) of eligible patients with a total of 90 lesions. The primary endpoint of in-stent late loss (LL) was 0.35±0.63 mm (95% CI: 0.22 to 0.49) and the in-segment LL was 0.15±0.35 mm (95% CI: 0.07 to 0.23). The LL frequency distribution is shown in Figure 1. The rate of in-stent binary restenosis was the same as in-segment and was 12.2% (11 lesions of 90) including two complete occlusions. QCA analysis results are summarised in Table 3.
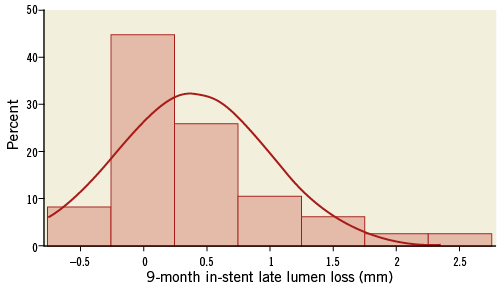
Figure 1. Frequency distribution of LL at nine-month angiographic follow-up.
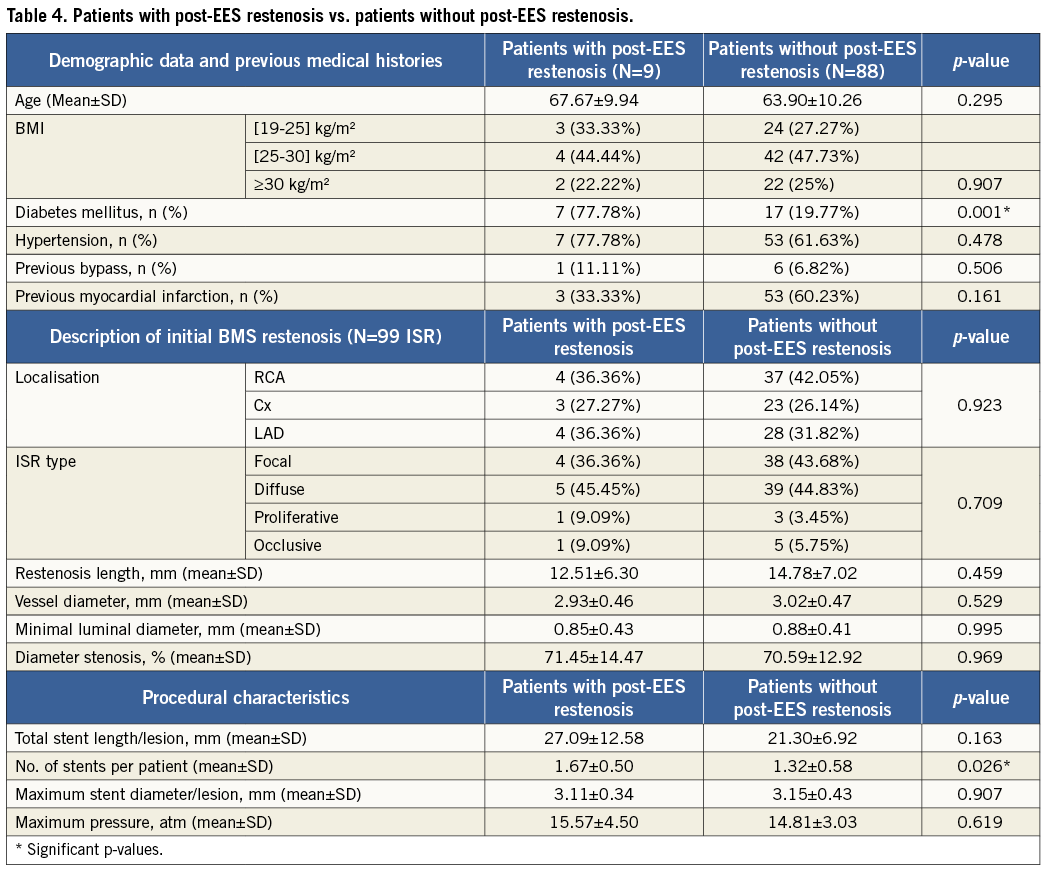
A comparison has been performed between XERES patients with post-EES restenosis and patients without post-EES restenosis. The results are summarised in Table 4.
INTRAVASCULAR ULTRASOUND EVALUATION
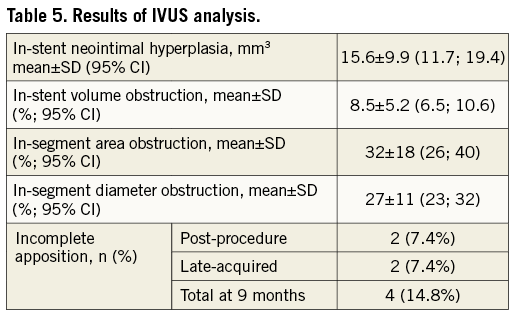
Volumetric intravascular ultrasound data were available in 27 lesions (26 patients or 96.3% of patients who initially underwent IVUS examination) at nine months. The average volume of neointimal hyperplasia was 15.6±9.9 mm3 or 0.66±0.44 mm3 per mm of stent length. The in-stent percent volume obstruction was 8.5±5.2%. The in-segment percent area and diameter obstruction were 32±17% and 27±11%, respectively. Four patients had a lumen area <4 mm². Two initial stent incomplete appositions were persistent and two other patients had late acquired incomplete apposition (Table 5).
MAJOR ADVERSE CARDIAC EVENTS
Major adverse cardiac events (MACE) at nine-month follow-up are listed in Table 6. Clinical follow-up was available for 100% of 97 patients. The cumulative incidence of MACE was 17.2%. Seven TLR, of which five were ischaemia-driven, were observed in five patients. One patient had a stent thrombosis during the initial procedure, treated by re-PCI. Five patients had periprocedural myocardial infarction and one patient had a myocardial infarction during the follow-up period. One 80-year-old patient had a sudden death while driving, five months after the initial procedure. This was classified as possible stent thrombosis (despite dual antiplatelet regimen) and cardiovascular death. No other death was observed. Time-to-event curve for TLR is shown in Figure 2.
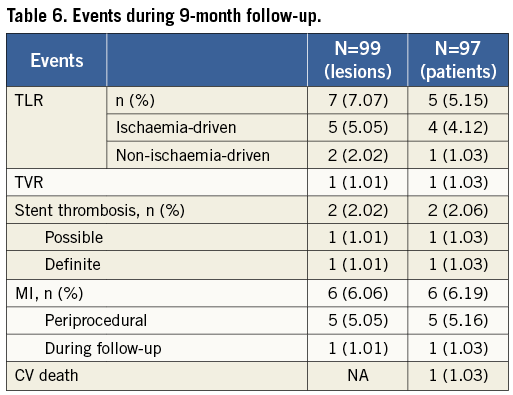
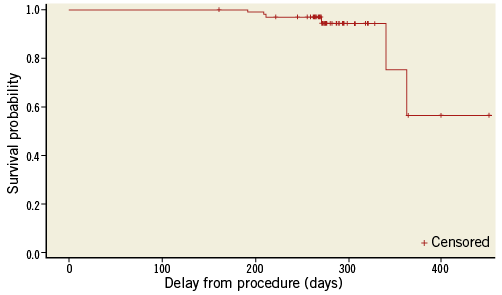
Figure 2. Time-to-event curve for TLR. Number of subjects: 97; Event: 7 TLR in 5 patients.
Discussion
The major finding of the present study is that EES implantation for in-stent restenosis is safe and associated with low recurrence rates of restenosis and acceptable LLL.
The population of this study is broadly comparable to the baseline patient and lesion characteristics found in the everolimus arm of a non-randomised comparative study on the use of drug-eluting stents in the treatment of in-stent restenosis (ISR)15. It is also comparable to that of a single-arm study, TAXUS III, which assessed the feasibility and safety of paclitaxel-eluting stents for the treatment of ISR17.
The rate of angiographic nine-month follow-up (86.3%) is similar to that of other studies that enrolled patients with recurrent in-stent restenosis17,18. This rate differs from the nine-month clinical follow-up rate (100%) due to seven patients refusing the angiographic control in the absence of symptoms. The XERES study included a population of patients with in-stent restenosis who had to undergo a significant number of procedures and exams, which may explain why some of them did not agree to undergo further angiographic control in the absence of symptoms.
The late loss observed at nine-month follow-up in the XERES study was 0.35±0.63 mm (95% CI: 0.22 to 0.49), a value intermediate between that reported six months after stenting with paclitaxel-eluting stent (PES) (0.54±0.51 mm)17, and sirolimus-eluting stent (SES) (0.17±0.76 mm)18. In PEPCAD II, a prospective, randomised, multicentre, two-arm phase II pilot study which examined the safety and efficacy of the SeQuent® Please drug-eluting balloon (DEB) (B. Braun Melsungen AG, Berlin, Germany), coated with a combination of paclitaxel and an x-ray contrast medium called iopromide, in the treatment of ISR in native coronary arteries for procedural success and preservation of vessel patency compared with the TAXUS™ DES (Boston Scientific, Natick, MA, USA)19, the LL reported was 0.19±0.39 mm (95% CI: 0.10 to 0.28)19. Note that these were LL values at six-month follow-up whereas the XERES study reported LL values at nine-month follow-up. Otherwise, 13 (14.4%) negative late loss values have been observed in the XERES study and the largest negative value observed was –0.86 mm, due to a coronary aneurysm formation. In the SPIRIT II trial10, a single-blind multicentre non-inferiority randomised controlled trial which evaluated the safety and performance of the XIENCE V everolimus-eluting stent (EES) versus the TAXUS paclitaxel-eluting stent (PES) in the treatment of patients with de novo native coronary artery lesions, the in-stent LL at six-month follow-up in the everolimus arm was 0.11±0.27 mm with 30% negative late loss value observations. The largest negative LL value was –0.57. It is important to note that LL is a parameter with a large standard deviation when inter-observer variability is assessed (1SD 0.36 mm, 2SD 0.72 mm)20 and it is a result of two individual measurements (MLD post-procedure and MLD at follow-up), both of which have their own inter-observer variability due mainly to the process of calibration21.
Binary restenosis rates were 14.6%, 16% and 7%, respectively, for SES18 PES17 and DEB19 versus 12.2% for everolimus in our study. Our results compare favourably with other DES despite the difference in follow-up time points (six months in TAXUS III and RESEARCH versus nine months in XERES). The comparisons between DES are presented in Table 7.
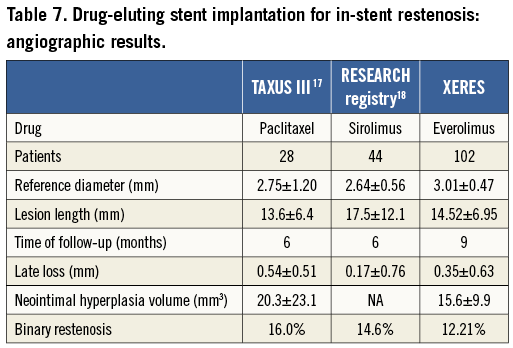
The outcomes of patients with in-stent restenosis after repeat treatment have been reported to be closely related to the baseline lesion characteristics. The risk profile increases progressively from lesions with a focal pattern to lesions with a more diffuse appearance and total occlusions16. However, there was no significant difference between patients with post-EES and those without post-EES restenosis concerning the initial ISR type in our study (p=0.709).
Otherwise, the outcomes of percutaneous coronary interventions are reported to be worse in patients with diabetes compared with those without diabetes22,23. In the drug-eluting stent arm of randomised trials24-26, the presence of diabetes has been reported to increase the risk of restenosis by 1.6 to 2.2-fold compared with the risk for non-diabetics. Our study confirms these findings since the univariate analysis shows that the presence of diabetes mellitus at baseline is correlated with post-EES restenosis at nine months. However, LL was not statistically significantly different between diabetic patients and patients without diabetes.
The nine-month follow-up cumulative MACE rate in the XERES study (17.2%) is favourable, compared to the six-month follow-up results in the TAXUS III trial (29%)17. In other DES trials, MACE rates observed were 4.6% versus 8.1% at nine-month follow-up, for EES and PES, respectively (relative risk, 0.56 [95% CI: 0.34 to 0.94]; p=0.03) in SPIRIT III12 and 5% in the EES group versus 8% in the PES group (p-value for superiority was 0.005) in COMPARE14. COMPARE was a randomised trial which compared the safety and efficacy of the EES and PES in real-life practice.
TLRs are the most common MACE reported. This can be explained by the fact that a systematic angiographic control was performed at nine-month follow-up. Indeed, as shown in the BENESTENT II trial, a systematic angiographic control can lead to a “flurry” of additional revascularisation procedures compared with clinical follow-up alone, although these revascularisations are also clinically driven. In this study, the incidence of repeat TLR was significantly greater (14.4% vs. 9.1%) in patients who underwent routine angiographic follow-up than in those who had clinical follow-up alone27. An expert consensus document of the American College of Cardiology confirms that the rates of revascularisation are higher in studies with mandated routine angiographic follow-up28.
Neointimal hyperplasia volume (15.6±9.9 mm3) was lower than that reported in the TAXUS trial (20.3±23.1 mm3)17. Comparable results have been reported in the randomised SPIRIT II and III trials, where the EES arms showed significantly lower neointimal hyperplasia volume and in-stent volume obstruction compared to the PES arms10,12.
The rate of stent malapposition (7.4% at the completion of the procedure and 7.4% at nine-month follow-up) was lower than that of the EES arm in the SPIRIT III trial (34.4% at the completion of the procedure and 25.6% at eight-month follow-up)12, where the IVUS examination was performed in 14.2% of 1,002 randomised patients. Thus, the better immediate post-procedural stent expansion in our study might explain the lower rate of late acquired malapposition.
The IVUS examination also made possible the determination of the lumen obstruction mechanism at follow-up. This was mainly due to incomplete stent expansion (74%), while neointimal proliferation was implicated in only 26% of cases. These IVUS results on stent malapposition and on the lumen obstruction mechanism highlight the importance of adequate stent deployment for the treatment of in-stent restenosis lesions.
The limitations of this study are its relatively small sample size and the single-arm open label design without randomisation. In all cases, the one-year and two-year clinical follow-up of patients will give us additional information on the long-term outcomes of this population.
Conclusion
This prospective, multicentre study shows the potential utility and safety of EES for the treatment of bare metal in-stent restenosis. The XERES IVUS substudy confirmed the good results achieved with XIENCE stent implantation for in-stent restenosis. Neointimal proliferation was very limited and three quarters of lumen narrowing at follow-up was due to suboptimal stent deployment with limited neointimal proliferation.
Conflict of Interest statement
The authors have no conflicts of interest to declare.
