
Since the advent of percutaneous coronary intervention (PCI) in the late 1970s, successive technological innovations have been essential to the observed improvement in patient outcomes with this technique. Nowadays, contemporary drug-eluting stents (DES) are characterised by excellent efficacy and safety profiles during long-term follow-up1. Specific design developments have contributed to the high performance, including the use of newer metallic alloys permitting thinner strut dimensions, biocompatible polymer coatings, and more effective antiproliferative agents. However, suboptimal performance is observed and remains a challenge in certain high-risk subgroups. For example, an unmet need may be said to exist for new devices in patients with diabetes, end-stage renal failure, or diffuse multivessel disease, or in lesions located at coronary bifurcations, in saphenous vein grafts, or in chronic total occlusions.
In this issue of EuroIntervention, Moreu and colleagues report a first-in-man evaluation of the novel durable fluoro-acrylate polymer-based sirolimus-eluting Angiolite stent (iVascular, Barcelona, Spain), which was directly compared with the durable fluoropolymer-based everolimus-eluting XIENCE stent (Abbott Vascular, Santa Clara, CA, USA) in patients with de novo coronary artery disease (CAD)2.
A total of 223 patients were randomly allocated in a 1:1 fashion to receive either device, in the setting of a multicentre trial. The trial was powered for non-inferiority for in-stent lumen loss (LLL). At six-month angiographic surveillance, LLL was 0.04±0.39 mm in patients treated with the Angiolite stent and 0.08±0.38 mm in those treated with the XIENCE stent (difference=–0.04 mm [95% confidence interval: –0.15 to 0.07], p non-inferiority=0.002). A subgroup of 88 patients underwent optical coherence tomography evaluation. These data suggested a lower neointimal thickness with the Angiolite stent than with the XIENCE stent (86.4 μm vs 72.1 μm; p<0.01), though how this should be interpreted in light of a lack of clear difference in LLL is an open question. There were no differences in terms of uncovered struts score (9.0% vs 9.9%, p=0.41).
Due to a number of high-profile device failure cases, regulatory processes for approving high-risk medical devices have been the subject of much scrutiny in the medical and lay media in recent years3,4. In Europe, a new medical device regulation (MDR 2017/745) was published and became law in May 2017; it will be fully implemented from May 20205. There are a number of important changes in terms of strengthening of requirements for clinical investigations for approval of high-risk devices, as well as in technical documentation, methods for device tracking, and transparency of approval processes.
By virtue of their placement in and contact with the circulatory system, coronary stents are classified as high-risk devices. When considering what type of evidence is sufficient for applications for approval of novel coronary stents, regulatory bodies refer to technical standards from organisations such as the International Standards Organization (ISO) and other device-specific guidance documents. In Europe, a device-specific guidance document for the evaluation of coronary stents exists and was published by the European Commission more than a decade ago6. A number of years ago, in response to a request from the European Commission, an ESC-EAPCI Task Force on Coronary Stents provided recommendations for a revision of this document1. The key recommendations were that only devices with satisfactory non-clinical assessment should undergo clinical evaluation. Moreover, initial human feasibility studies should be small-sized (N=50-150) and performed in selected patients – a single-arm, prospective observational design may be appropriate – followed by subsequent medium-sized randomised trials (N=200-500), powered for detecting differences in surrogate endpoints, compared against contemporary DES and incorporating intracoronary imaging in a subgroup of patients. Finally, a large-scale clinical trial with broad inclusion criteria and long-term follow-up should be carried out after approval.
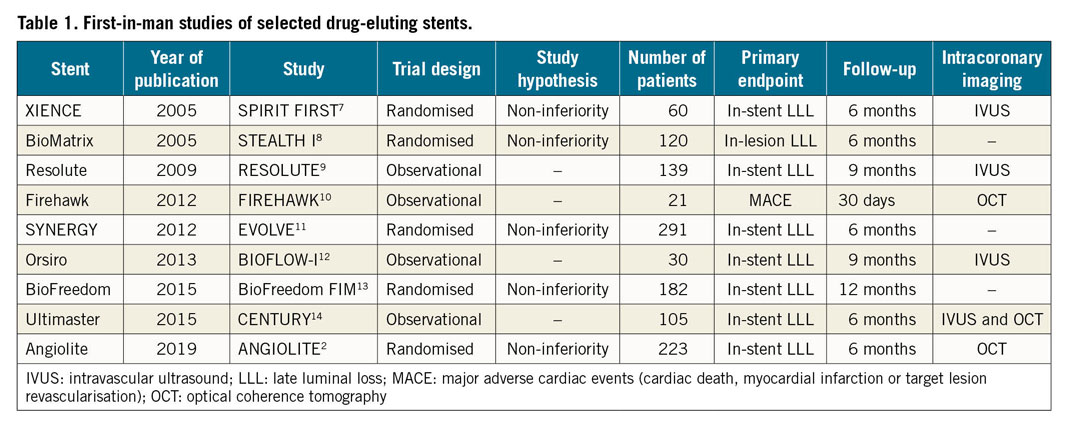
The study of Moreu and colleagues represents a first-in-man evaluation of a novel DES technology in the setting of a randomised controlled trial based on a well-established surrogate primary endpoint. DES is a mature technology in terms of trial data, and benchmark values for mean late lumen loss in these types of trial are well known. The data presented suggest that the investigational device has an antirestenotic performance in line with already approved devices. The type of evaluation is well aligned with recommendations such as those outlined in the Task Force report and elsewhere. Interestingly, first-in-man studies of new DES have historically been carried out in a relatively small number of patients, often without an active control, and without a broad use of intracoronary imaging (Table 1). This means that information on comparative effectiveness is limited7,8,9,10,11,12,13,14. Studies such as those of Moreu and colleagues generate data well suited for decisions on regulatory approval and are a welcome development in the field. However, subsequent large-scale clinical trials are critical to confirm the results. These larger trials will provide more information on low frequency adverse events such as stent thrombosis and clinical performance in less selected patients. This is essential in order to investigate whether this new device will represent a valuable addition to the armamentarium of the practising interventional cardiologist.
Conflict of interest statement
R.A. Byrne reports lecture fees from B. Braun Melsungen AG and Biotronik, outside the submitted work. G.G. Stefanini reports a research grant (to the institution) from Boston Scientific, and speaker/consultant fees from B. Braun, Biosensors, Boston Scientific, and GADA, outside the submitted work. The other author has no conflicts of interest to declare.
