
Abstract
Aims: The durable fluoroacrylate polymer-based sirolimus-eluting stent (Angiolite SES) has shown promising preclinical and clinical results regarding inflammatory vascular reaction and neointimal healing. We aimed to compare performance between the Angiolite SES and an everolimus-eluting stent (EES) in patients with coronary artery disease.
Methods and results: The ANGIOLITE trial, a prospective, randomised, multicentre trial, compared the restenosis parameters of both stents in de novo coronary lesions. The primary endpoint was late lumen loss at six-month angiographic follow-up. In-stent healing was assessed by optical coherence tomography (OCT). The main clinical endpoint was target lesion failure (TLF) evaluated up to 24 months. A total of 223 patients were randomised 1:1 to EES or SES. At six months, in-stent late lumen loss was 0.08 mm (±0.38) for EES vs 0.04 mm (±0.39) for SES (difference=–0.04 mm, 95% CI: –0.15, 0.07, p for non-inferiority=0.002). By OCT, the rate of uncovered to total number of struts score >30% was comparable between the groups whereas neointimal thickness was reduced in the SES arm (9.0% [7.6, 10.6] vs 9.9% [8.5, 11.3], p=0.41; and 86.4 [81.6, 91.2] µm vs 72.1 [68.2, 76.0] µm, p<0.01, respectively). At 24 months, TLF occurred in eight patients (7.6% [3.3, 14.5]) in the EES arm and in seven patients (7.1% [2.9, 14.0]) in the SES arm (p=0.88). The definite/probable stent thrombosis rate was comparable between the groups (1.9% [0.2, 6.7] vs 1.0% [0.0, 5.5] EES vs SES, respectively; p=0.59).
Conclusions: This trial demonstrates similar antirestenotic efficacy at midterm follow-up of the Angiolite SES vs an EES. Clinical endpoints were comparable between the groups at two-year follow-up.
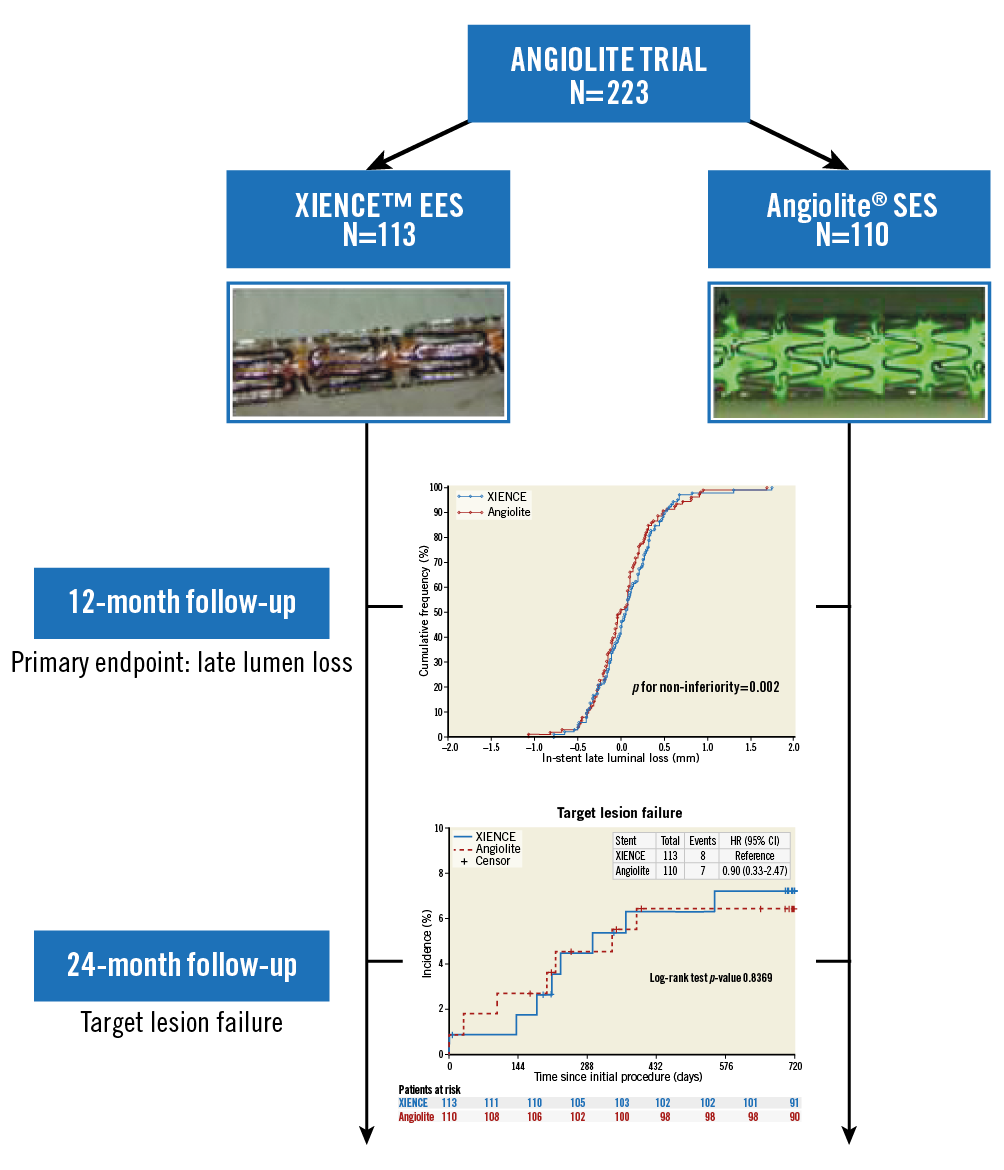
Visual summary. Main results of the ANGIOLITE trial.
Introduction
The design of drug-eluting stents (DES) with an antiproliferative drug included in a polymer matrix was a breakthrough in the field of percutaneous coronary intervention (PCI), leading to a significant reduction of major adverse cardiac events (MACE) that was mainly driven by a lesser number of new revascularisations1,2. However, a higher than expected rate of stent thrombosis was observed with first-generation DES, which caused health alerts3 and forced the medical community to recommend prolonging the dual antiplatelet regimen4. The permanent polymer used in those early stents was responsible for chronic inflammation, delayed endothelialisation, and stent thrombosis5. New-generation DES incorporated several modifications, such as the use of biocompatible polymers, different antiproliferative limus analogues and metallic alloys allowing the design of thinner struts, and modifications in stent architecture. As a result, they offered similar antirestenotic efficacy but a better safety profile, compared to their first-generation counterparts. Among second-generation DES, the cobalt-chromium everolimus-eluting stent (EES) has shown favourable efficacy and safety profiles and has been used as a workhorse in routine clinical practice and as a control arm in many randomised trials evaluating the performance of new DES6.
The Angiolite® stent (iVascular, Barcelona, Spain) is a thin-strut cobalt-chromium sirolimus-eluting stent (SES) with an open-cell design containing a durable biostable coating composed of three layers - acrylate to ensure adhesion to the metal surface, fluoroacrylate that carries the sirolimus (1.4 microgr/mm2), and a top layer of fluoroacrylate to control drug release (>75% elution within the first month). This composition demonstrated in vitro early endothelial cell growth and a reduction of smooth muscle cell proliferation (Supplementary Appendix 1, Supplementary Table 1, Supplementary Table 2, Supplementary Figure 1-Supplementary Figure 5). Preclinical studies in animals have demonstrated a favourable healing process with a reduction in injury score and inflammation score that led to a reduction in neointimal area and an increase in the percentage of endothelialised surface as compared to the EES stent7. These preclinical results were later confirmed in the ANCHOR trial8 that assessed strut healing after Angiolite SES implantation. As early as three months after implantation, the percentage of strut coverage was nearly 90%. For these reasons, we considered the design of a non-inferiority trial against the world’s most commonly implanted DES. (www.clinicaltrials.gov NCT03049657).
Methods
PATIENTS AND STUDY DESIGN
This prospective, randomised, multicentre, controlled trial was designed to test the non-inferiority of the Angiolite SES in comparison with EES in patients with coronary artery disease. Detailed inclusion and exclusion criteria are presented in Supplementary Appendix 2. Briefly, patients aged at least 18 years, with ischaemic heart disease (stable angina, silent ischaemia, or acute coronary syndrome) and scheduled for PCI of de novo epicardial coronary stenosis were eligible. Patients were enrolled at 11 academic medical centres in Spain between February 2016 and February 2017 (Supplementary Table 3). Written informed consent was obtained from each patient prior to study enrolment. All participants were randomly allocated in a 1:1 ratio to either the SES or EES. To generate comparable groups regarding known and unknown risk factors, randomisation was independently conducted online via a web-based application. The randomisation was balanced and stratified by participating centre and allocated treatment group. All centres received the approval of their medical ethics committee. The study was conducted in compliance with the protocol, the Declaration of Helsinki, BS EN ISO 14155 Part 1 and Part 2, and applicable local requirements. A description of the data and safety monitoring board and clinical events committee can be found in Supplementary Table 4.
STUDY ENDPOINTS AND DEFINITIONS
The primary endpoint of the trial was in-stent late lumen loss (LLL), defined as the angiographic minimum lumen diameter (MLD) immediately after PCI minus the MLD at angiographic follow-up at six months measured by off-line quantitative coronary angiography (QCA) within in-stent boundaries. Additionally, as an exploratory analysis, we aimed to compare the rate of target lesion failure (TLF) as a composite of cardiac death, target vessel-related MI or clinically driven target lesion revascularisation (TLR) at 12 months. Secondary clinical endpoints included device success, procedural success, MACE (a composite of all-cause death, any MI or any revascularisation) and stent thrombosis defined according to the Academic Research Consortium criteria9. The main secondary angiographic endpoints included acute gain, in-segment LLL, MLD, percentage diameter stenosis and binary restenosis (Supplementary Appendix 3). An optical coherence tomography (OCT) study was performed at six-month follow-up in a cohort of patients in five predefined centres (OCT subgroup). The OCT parameters included the OCT-derived stent-level and strut-level neointimal proliferation, strut coverage measured by the % of uncovered stent struts and the number of cross-sections by rate of uncovered to total number of struts (RUTTS) score >30%, and rates of incomplete stent apposition (Supplementary Appendix 4). Clinical follow-up was scheduled at 1, 6, 12 and 24 months.
CORONARY STENTING PROCEDURE
Coronary interventions were performed according to current standard techniques. All patients received aspirin (300 mg) and a loading dose of clopidogrel (600 mg) or ticagrelor (180 mg) prior to the procedure, unless already receiving these drugs. Heparin i.v. was given to maintain an activated clotting time at >250 sec with an additional bolus during the procedure if needed. After the procedure, aspirin was prescribed indefinitely (100 mg/day), and clopidogrel (75 mg/day) or ticagrelor (90 bid) or prasugrel (10 mg/day) was prescribed for a minimum of six months after the index procedure. Specific descriptions of the stents used in this trial are presented in Supplementary Table 1 and Supplementary Table 2.
STATISTICAL ANALYSIS
To test the non-inferiority of the Angiolite SES versus the XIENCE EES (Abbott Vascular, Santa Clara, CA, USA) in terms of six-month in-stent LLL, a mean late loss of 0.10 mm (standard deviation [SD] 0.45 mm) was assumed for both stents, which was extrapolated from the QCA results for the EES in the SPIRIT I and SPIRIT II trials10,11. For non-inferiority testing, with a 0.2 mm non-inferiority margin, type I error at 0.05 (one-sided), 90% statistical power, and 1:1 sampling ratio (Angiolite SES: EES), a sample of 176 patients (88 per group) was needed. Assuming a 12% dropout rate during follow-up, a total of 200 patients (100 per group) constituted the final calculated sample size. We finally decided to increase this sample size by 10% to ensure the inclusion of at least 80 patients in the OCT substudy.
For continuous variables, results are presented as mean±SD. Variables were compared using an independent t-test or the Mann-Whitney test when applicable. Categorical variables are presented as counts and percentages and compared using the chi-square test or Fisher’s exact test. The null hypothesis was evaluated on a non-inferiority basis, using a mixed effects linear regression model of the mean in both groups. Angiographic and OCT outcomes were analysed at lesion level with mixed effects linear regression models (continuous variables) or mixed effects logistic regression models (categorical variables) that account for the non-independence of multiple lesions within patients. Clinical variables at 12 and at 24 months were compared with the χ² test. Time-to-event hazard curves, presented with Kaplan-Meier estimates, were compared using a log-rank test. Associations were considered statistically significant in the presence of a two-sided p-value <0.05.
Results
PATIENTS AND PROCEDURES
A total of 223 patients were enrolled in the trial: 110 were allocated to SES and 113 to EES (Figure 1). The two groups were well balanced in terms of baseline clinical characteristics (Supplementary Table 5). Mean age was 63.0 years, with a male preponderance (78.5%). More patients in the EES group had prior myocardial infarction (MI) (16.1% vs 7.3%; p=0.04) and prior revascularisation procedures (18.8% vs 9.1%; p=0.04). There were differences in lesion type distribution (p=0.02), with more type C lesions in the SES group (3.3% vs 9.5%). Device success was achieved in 99.3% of lesions in the SES group and 100% in the EES arm. Procedural success was achieved in 99.3% of lesions in both arms (Table 1).
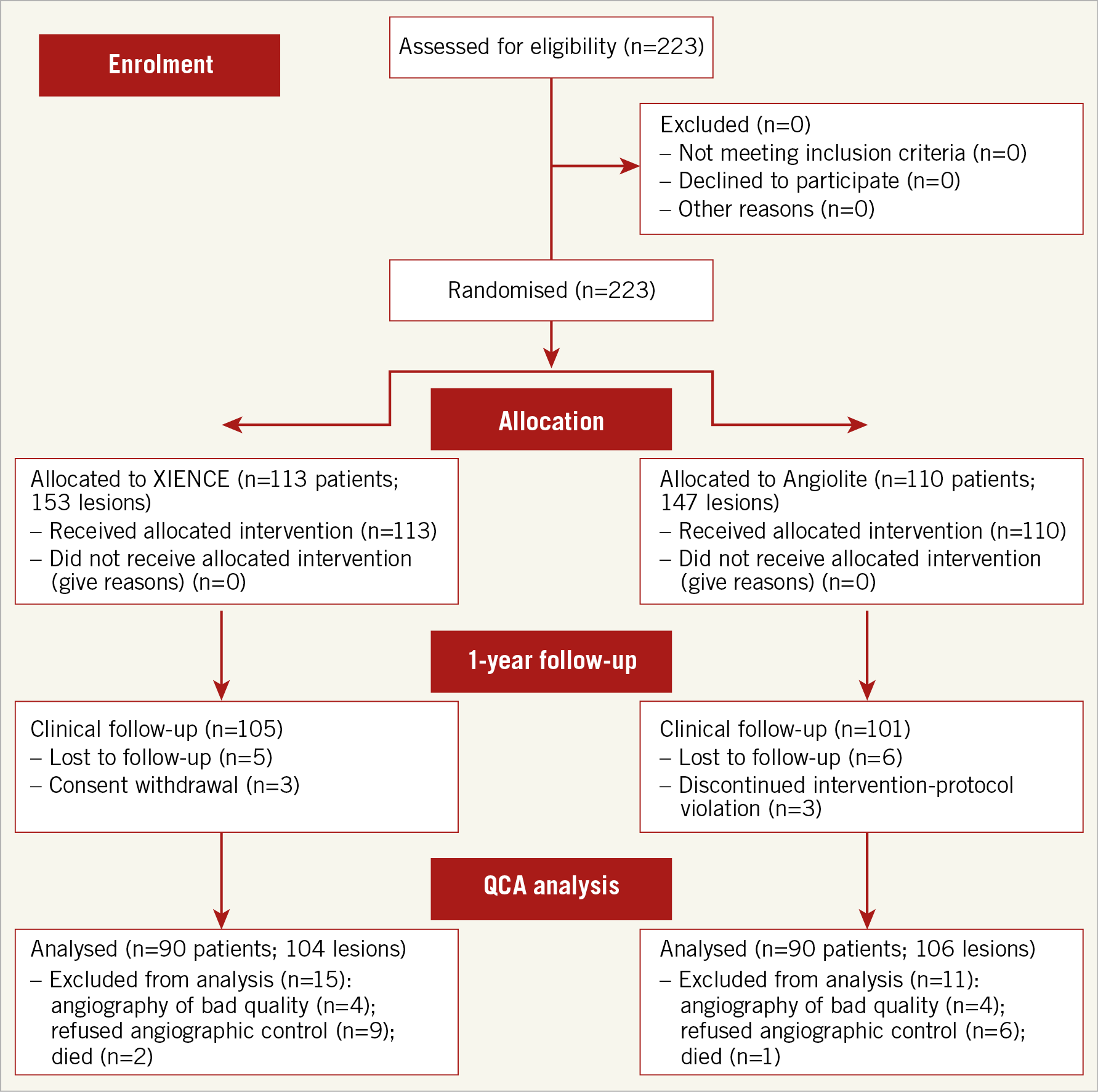
Figure 1. Flow chart of the study according to CONSORT 2010 guidelines.
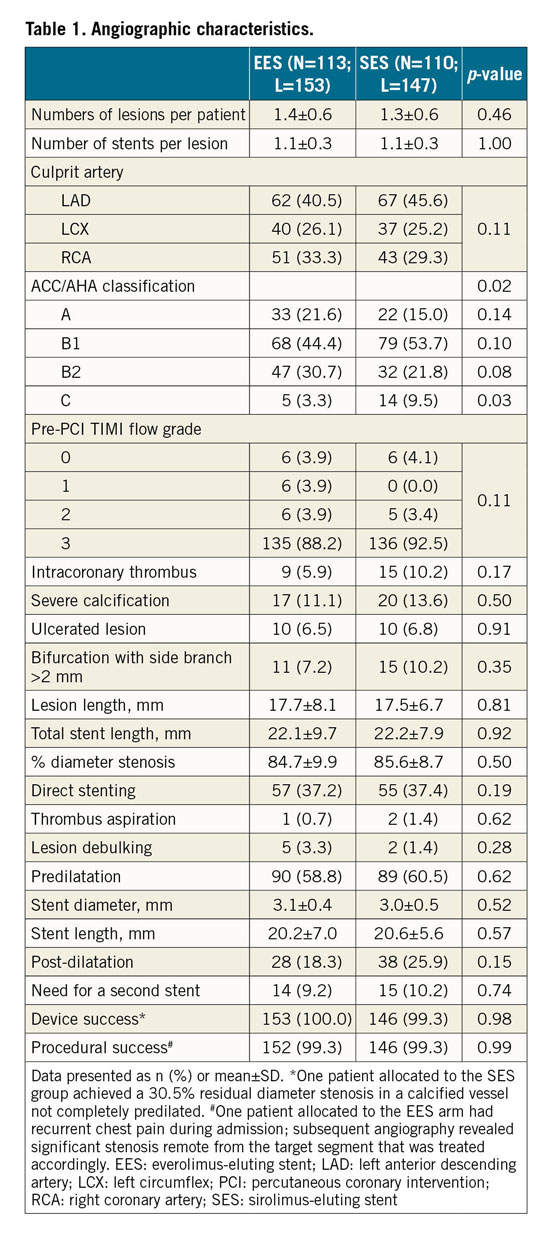
QUANTITATIVE CORONARY ANGIOGRAPHY RESULTS
Baseline and post-procedure QCA data were similar between the groups (Table 2). The acute gain in the SES group was 1.65±0.48 mm versus 1.64±0.50 mm in the EES group (p=0.84). Follow-up angiography was performed in 90 patients (106 lesions) in the SES group (81.8% of those allocated) and in 90 patients (104 lesions) in the EES group (79.6%). The primary endpoint, in-stent LLL, was 0.04±0.39 mm in the SES group and 0.08±0.38 mm in the EES arm (difference=–0.04 mm, 95% CI: –0.15, 0.07, p for non-inferiority=0.002) (Figure 2). Similarly, in-segment LLL was non-inferior between the groups (0.00±0.44 mm in the SES group vs 0.06±0.38 mm in the EES group; difference=–0.06 mm, 95% CI: –0.18, 0.06, p for non-inferiority=0.007). Cumulative frequency distributions of in-stent and in-segment LLL curves are presented in Figure 2A and Figure 2B. In-stent binary restenosis occurred in three patients, two in the EES group and one in the SES arm. Cumulative frequency distribution curves of MLD pre intervention, post intervention and at follow-up are presented in Supplementary Figure 6.
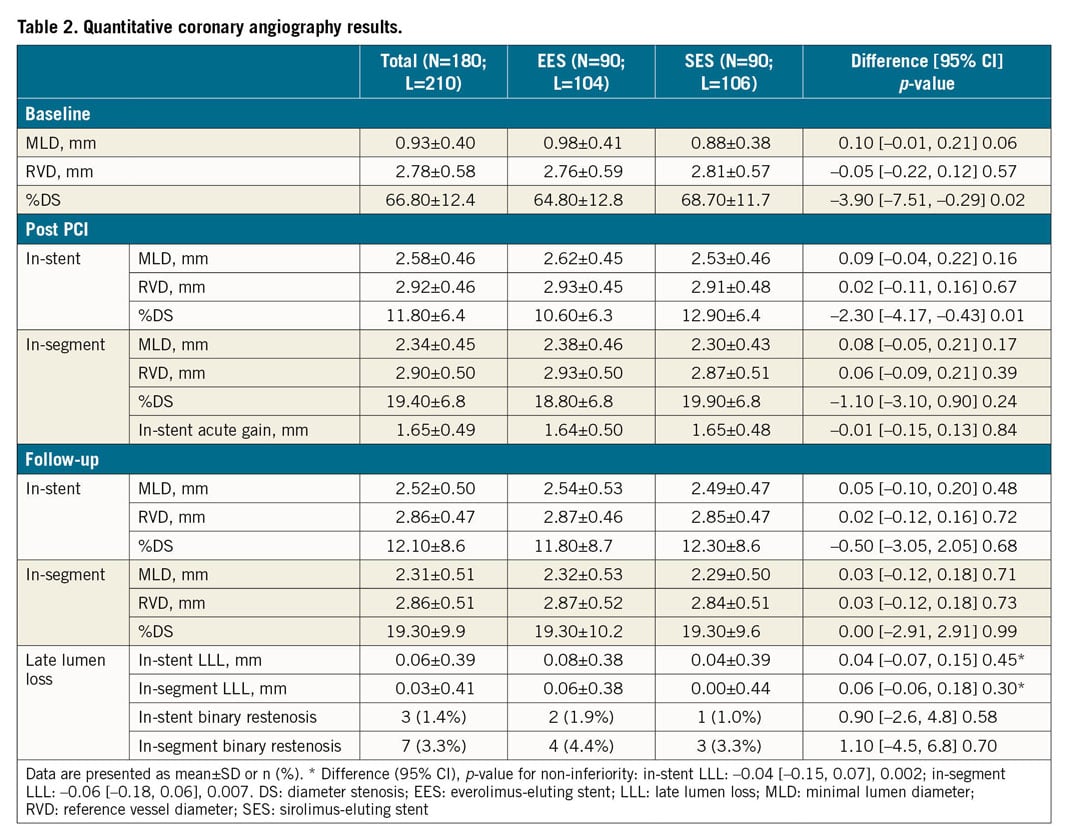
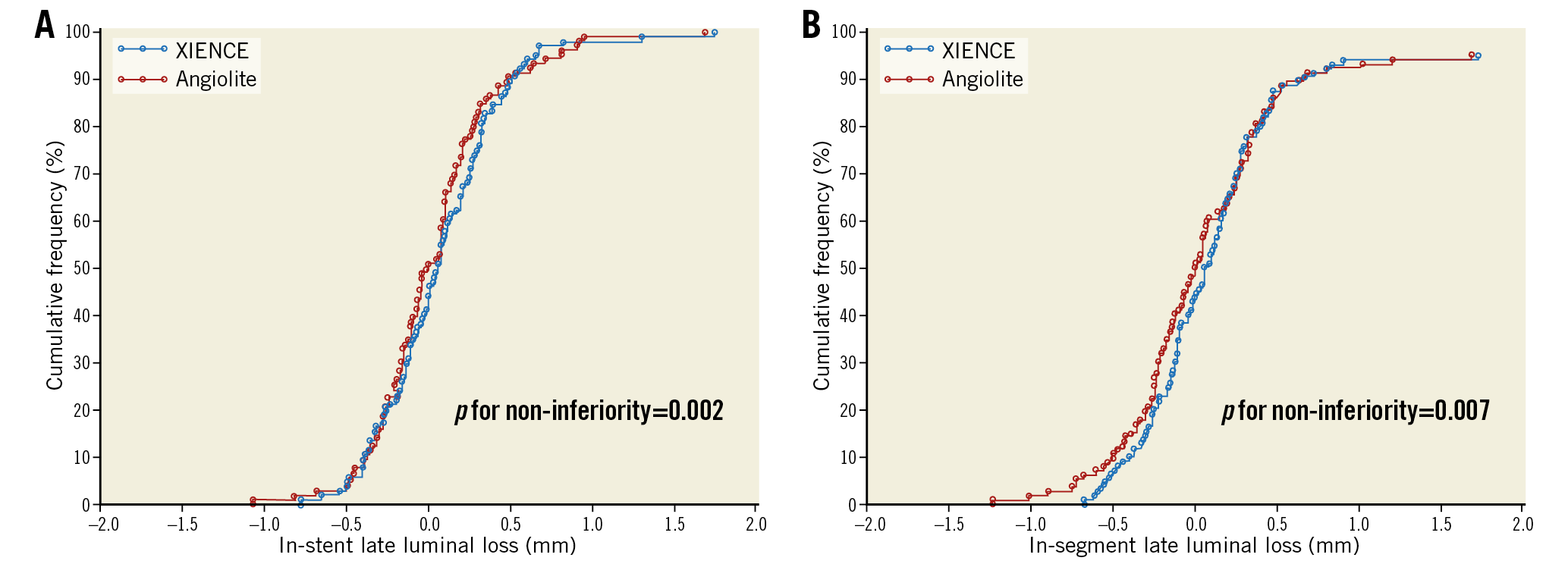
Figure 2. Cumulative frequency distribution curves. A) In-stent LLL. B) In-segment LLL.
OPTICAL COHERENCE TOMOGRAPHY SUBSTUDY
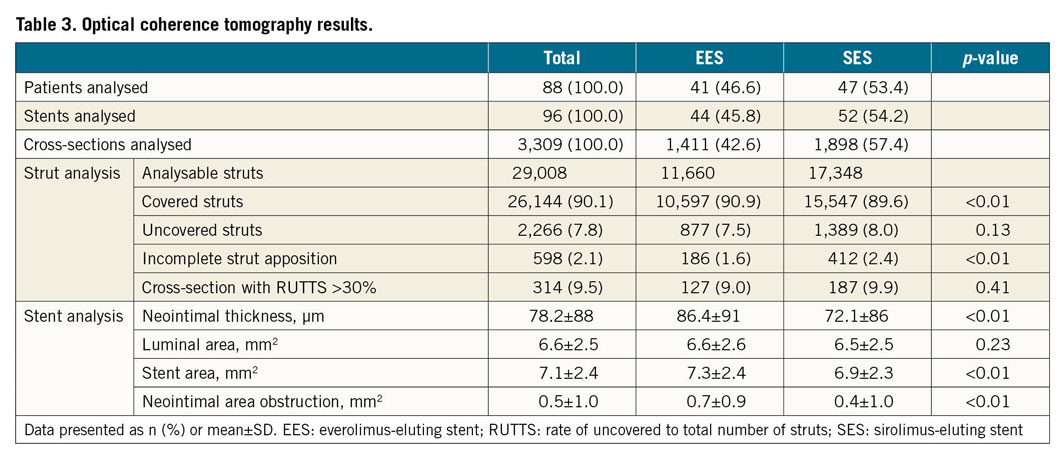
A total of 88 patients were included in the OCT substudy (47 in the SES group and 41 in the EES group). There were no differences between the groups in baseline characteristics (Supplementary Table 6). The main QCA parameters in this subgroup of patients mimicked those of the overall study population (Supplementary Table 7).
The main OCT findings are displayed in Table 3. In the EES arm, 10,597 struts (90.9%) were fully covered, RUTTS score >30% was observed in 9.0% of the analysed cross-sections and 1.6% of struts were incompletely apposed. In the SES arm, 15,547 struts (89.6%) were fully covered, the RUTTS score >30% was observed in 9.9% of analysed cross-sections, and 2.4% of struts showed incomplete apposition. Mean neointimal thickness and neointimal area obstruction were lower in the SES group.
CLINICAL OUTCOMES
During hospitalisation, there were no differences in clinical outcomes between the groups. Three major complications prolonging hospital stay occurred – one acute definite stent thrombosis in the EES group and two bradyarrhythmia events not related to the device in the SES arm.
At one year, there were no differences in outcomes between the groups (Figure 3, Supplementary Table 8). Eleven patients presented with TLF (seven in the EES group and four in the SES group). In the EES arm, this included one cardiac death at seven months due to cardiac arrest at home, two acute MI (one in the first 24 hours post PCI due to stent thrombosis and the other at 7.1 months), and four clinically driven TLRs. The SES arm had one MI secondary to definite stent thrombosis at 7.4 months and three clinically driven TLRs. Additionally, there were two non-cardiac deaths (one secondary to colonic necrosis in the EES arm and the other secondary to staphylococci meningitis in the SES group) and nine non-target vessel revascularisations (three in the EES arm and six in the SES group).
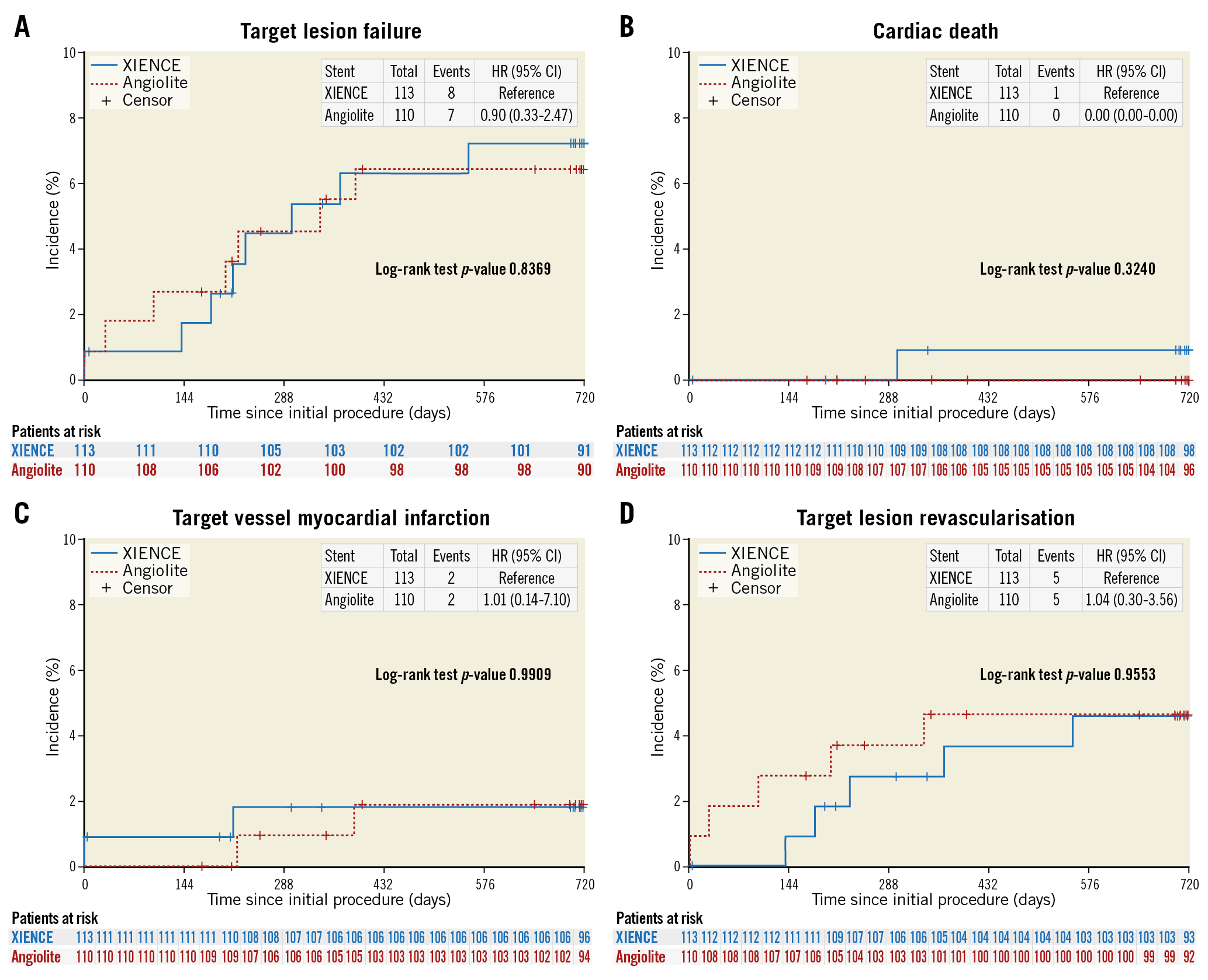
Figure 3. Kaplan-Meier curves. A) TLF. B) Cardiac death. C) Target vessel myocardial infarction. D) TLR.
Final clinical follow-up at 24 months was obtained in 91.5% of patients. Most patients in both groups had discontinued dual antiplatelet therapy (DAPT) (21% remained on DAPT in the EES arm vs 21.1% in the SES arm; p=0.96) (Supplementary Table 8). Overall, there were no differences in outcomes between the groups (TLF 7.6% [3.3, 14.5] in the EES arm vs 7.1% [2.9, 14.0] in the SES arm; p=0.88). Concomitant medication was also comparable between the groups during follow-up (Supplementary Table 9).
Discussion
The ANGIOLITE study is the first prospective, multicentre, randomised controlled trial designed to assess the efficacy of the new second-generation Angiolite SES. As compared to the XIENCE EES, the SES appeared to be non-inferior in angiographic parameters of restenosis. From the clinical point of view, few events were observed in either group at two-year follow-up.
In-stent LLL is frequently used to quantify the degree of neointimal hyperplasia, as it reflects the biological activity occurring after stent implantation. It is a simple measure, easy to understand and rather intuitive, and has been used in several randomised controlled trials to compare the efficacy of different types of DES. Moreno et al12 concluded, using meta-regression techniques, that the number of patients needed to treat to reduce one TLR compared with a bare metal stent is related to late loss. In a recent patient-level meta-analysis of seven randomised clinical trials involving 2,426 patients, Asano et al13 reported an exponential relationship between in-stent LLL and the two-year incidence of TLR.
QCA provided very low mean values of LLL, especially in the measurements obtained in-segment that could be related to positive remodelling of the vessel after stent implantation. At the stented segment, mean LLL in the Angiolite SES group was similar to that obtained in the ANCHOR trial8 (0.04±0.36 mm vs 0.07±0.37 mm). The mean LLL in the EES group was also very low, in concordance with values reported in other trials14,15,16. In general, reported LLL values in current-generation DES are below a 0.20 mm threshold. The BIOFLOW-II trial15 compared EES to SES with biodegradable polymer drug release, showing a similar in-stent LLL between groups (0.10±0.32 mm vs 0.11±0.29 mm, respectively). The PRISON IV trial also showed very low LLL in the XIENCE arm (0.07±0.46 mm)16. Finally, LLL of the Resolute™ stent (Medtronic, Minneapolis, MN, USA) was also very low (0.14±0.37 mm) in the PIONEER trial17. Variations in LLL values across trials can be related to variability of core lab analyses and different timing of the angiographic follow-up (6 vs 9 months). A broad SD of LLL is typically seen when using DES. The important suppression of neointimal proliferation (with values of LLL close to 0) leads to a wide SD induced for the few restenoses that may occur. Besides, LLL after bare metal stent implantation used to show normal distribution as opposed to that after DES implantation, which is usually non-normal18. In such a scenario, comparison of medians could be more accurate. However, for historical reasons and comparability with previous stent generations, current DES trials ultimately keep using mean±SD for LLL comparisons. Overall, LLL of metallic coronary stents reflected the capacity to prevent neointimal proliferation, with early and late constrictive remodelling being prevented by the metallic backbone of the device. However, in the era of first-generation DES, eradication of neointimal proliferation was related to late events such as stent thrombosis5. Therefore, the analysis of LLL represents a surrogate of the antirestenotic efficacy, but it may not be enough to discriminate the quality of the healing process as a parameter of device safety. To that end, the concomitant use of imaging techniques such as OCT can be helpful. Indeed, a very low LLL may reflect an incomplete healing process with uncovered and malapposed struts, only seen on OCT19. Therefore, the findings of the current OCT substudy are reassuring and support the good safety profile of the Angiolite SES. In the previous ANCHOR study8, the healing process of the Angiolite SES stent was evaluated by OCT at three and six months after implantation. As early as three months, strut coverage was evident in 86.3% of struts and the incomplete apposition rate was 1.3%. In keeping with these data, the OCT substudy of the ANGIOLITE trial corroborated a high degree of SES strut coverage at six months (nearly 90%) with a low incomplete apposition rate.
From the clinical point of view, the number of events at two years was very low in both groups, reflecting good clinical performance without the occurrence of late catch-up events after discontinuation of DAPT.
Study limitations
Several caveats of the ANGIOLITE study warrant consideration. First, the study was designed to evaluate efficacy in terms of LLL, and therefore the sample size is too small to draw conclusions on clinical events. Second, we excluded patients with left main disease, ST-segment elevation MI Killip III-IV or total chronic occlusion, and therefore conclusions cannot be applicable to these specific groups. Third, OCT was performed in five pre-selected centres. However, baseline characteristics and outcomes of patients included in the OCT cohort were similar to those of the non-OCT cohort (Supplementary Table 10). Besides, OCT was only performed at six-month angiographic follow-up. Therefore, we cannot discern whether the incomplete stent apposition observed at follow-up was also present at baseline. Finally, a large-scale trial powered for clinical events with longer follow-up is needed to confirm the results of this study.
Conclusions
In conclusion, this first randomised trial with a novel thin-strut, cobalt-chromium SES with a durable fluoroacrylate-based biostable polymer found it to be non-inferior to the gold standard second-generation EES in terms of the angiographic parameters of restenosis.
|
Impact on daily practice The angiographic results and neointimal stent coverage of the novel Angiolite SES appeared to be comparable to those of the gold standard EES in a broad spectrum of coronary artery disease patients. The Angiolite SES can be incorporated as a good option in the armamentarium of the interventional cardiologist. |
Acknowledgements
We are grateful to Dr Ana Serrador for her work as supervisor of the core lab. We are also grateful to Mr José Montes from Effice SL for his work on the database and statistical analysis.
Funding
The trial was designed by the principal investigators and received an unrestricted grant from Cardiva 2. Effice SL was responsible for study management, data monitoring and statistical analysis. The grant provider was not involved in the conduct of the trial, data management, data analysis, or drafting of the manuscript, and did not participate in the decision to submit the manuscript for publication. The principal investigators had full access to the study data. The corresponding author had full responsibility for the decision to submit the report for publication.
Conflict of interest statement
J. Moreu has received honoraria from Biosensors, Boston, Cardiva, Edwards Lifesciences and Medtronic. R. Moreno-Gómez has received honoraria from Abbott, Astra, Amgen, Bayer, Biosensors, Biotronik, Boston, Cardiva, Daiichi Sankyo, and Medtronic. A. Pérez de Prado reports grants and personal fees from iVascular, personal fees from Boston Scientific, Cardiva, and Terumo, grants from Abbott, grants and personal fees from AstraZeneca, and grants from Medtronic, during the conduct of the study. The other authors have no conflicts of interest to declare.
