
Abstract
Aims: The aim of the study was to compare the safety and efficacy of the platinum-chromium-based everolimus-eluting stent (EES) with a cobalt-chromium EES.
Methods and results: We performed a prospective, multicentre, single-blind non-inferiority all-comers study randomising patients with stable or unstable coronary artery disease (2:1) to treatment with the platinum-chromium EES (n=1,952) or the control cobalt-chromium EES (n=1,028) in Europe (PLATINUM PLUS trial). The primary endpoint was target vessel failure (TVF) at 12 months, a composite of target vessel-related cardiac death, myocardial infarction (MI), and ischaemia-driven target vessel revascularisation (TVR). Among 2,980 patients, 33% presented with acute coronary syndromes, and 48% with multivessel disease. At 12 months, the intention-to-treat analysis determined that the platinum-chromium EES was non-inferior to the cobalt-chromium EES for the primary endpoint (86 [4.6%] patients vs. 32 [3.2%], absolute difference 1.4%, 95% confidence interval [CI]: –0.1-2.9; upper limit of the one-sided 95% CI: 2.57%; non-inferiority p=0.012; superiority analysis: hazard ratio [HR] 1.44, 95% CI: 0.96-2.16, p=0.08). In the per protocol analysis, however, the primary endpoint was significantly more common in the platinum-chromium EES (HR 1.64, 95% CI: 1.05-2.55, p=0.03). There were no significant differences in the rates of cardiac death (1.1% vs. 1.0%, p=0.78), MI (1.6% vs. 0.8%, p=0.09), or ischaemia-driven TLR (2.0% vs. 1.6%, p=0.49). The rates of ARC definite or probable stent thrombosis were comparable between platforms (0.8% vs. 0.5%, p=0.44).
Conclusions: At one year, the platinum-chromium EES satisfied the pre-specified criteria for non-inferiority relative to the control cobalt-chromium EES in this all-comers trial. (ClinicalTrials.gov number: NCT01342822)
Introduction
Ever-improving coronary stent technology has yielded improved clinical outcomes for patients undergoing percutaneous coronary intervention (PCI). In particular, the cobalt-chromium everolimus-eluting stents (EES) (PROMUS™; Boston Scientific, Marlborough, MA, USA, or XIENCE V®; Abbott Vascular, Santa Clara, CA, USA) have demonstrated a reduced incidence of stent thrombosis, myocardial infarction (MI), and restenosis compared to the first-generation paclitaxel-eluting stents (PES)1-3. More recently, a new-generation platinum-chromium EES (PROMUS Element™; Boston Scientific) has been designed. This novel metal alloy and redesigned scaffold architecture aims to improve the acute performance of the stent further by enhancing deliverability, radial strength, vessel conformability, and radiopacity4,5. This novel platform carries the same antiproliferative drug and biocompatible durable polymer as its cobalt-chromium predecessor6,7.
To date, the platinum-chromium EES has demonstrated similar safety and efficacy to the cobalt-chromium EES (XIENCE V) and the cobalt-chromium zotarolimus-eluting stent (ZES)8,9. The comparison between the platinum-chromium EES and the cobalt-chromium EES was, however, a rigorously controlled randomised trial of relatively low-risk patients. Thus, there remains a paucity of evidence comparing these two EES platforms in real-world clinical practice where more than half (55%) of all patients have off-label characteristics10,11. An all-comers trial design, with wide-ranging inclusion and few exclusion criteria, further improves the generalisability of the study results and affords the opportunity to assess the impact of low-frequency, clinically important differences between devices. Such studies are of particular relevance following the publication of recommendations from the Circulatory System Devices Advisory Panel of the US Food and Drug Administration12.
We sought to determine the safety and effectiveness of the platinum-chromium EES in an unrestricted population undergoing PCI compared to the cobalt-chromium EES (XIENCE PRIME®; Abbott Vascular).
Methods
STUDY DESIGN AND PATIENTS
We performed a multicentre, non-inferiority trial in 48 centres in Europe (ClinicalTrials.gov number: NCT01342822): France (17 centres), Germany (five centres), Italy (six centres), Macedonia (one centre), The Netherlands (one centre), Spain (10 centres), Switzerland (one centre), and the United Kingdom (seven centres).
Patients aged ≥18 years of age, undergoing PCI for stable coronary artery disease or acute coronary syndromes (non-ST-elevation and ST-elevation MI) were eligible for study inclusion if they had at least one coronary artery lesion amenable to stent implantation, of ≥50% diameter stenosis, in a vessel of reference diameter ≥2.25 and ≤4.25 mm by visual estimation. Single or multiple coronary artery or saphenous vein graft lesions were suitable, and no restrictions on lesion length or complexity were stipulated. Patients were not excluded on the basis of comorbid medical conditions, except where a concurrent illness could result in non-compliance with the study protocol or dual antiplatelet therapy, or could limit life expectancy to ≤1 year. Further exclusion criteria included: left ventricular ejection fraction ≤20%; known hypersensitivity or contraindication to aspirin, heparin/bivalirudin, clopidogrel/ticlopidine, prasugrel, platinum-chromium alloy, everolimus, or contrast media; pregnancy; participation in another investigational drug or device study; and an inability to provide informed consent. The study complied with the Declaration of Helsinki and was approved by the ethics committees of all enrolling institutions. All patients provided written, informed consent to trial participation.
Randomisation was performed centrally, after diagnostic coronary angiography, using an interactive internet-based allocation service. The list of treatment allocation was computer-generated and stratified according to the enrolling centre. Patients were randomly allocated on a 2:1 basis to treatment with the PROMUS Element or the XIENCE PRIME EES. The operators were aware of the assigned study stent during the index PCI, but the patients and clinical staff involved in follow-up care were blinded to the stent allocation. Clinical follow-up by office or telephonic interview was scheduled at 30 days, one and two years after the index intervention.
PROCEDURES
All PCI procedures were performed according to local standard techniques. The recommended antiplatelet pre-treatment was 75-325 mg of aspirin, combined with a 600 mg loading dose of clopidogrel (if clopidogrel naïve), or a 60 mg loading dose of prasugrel. Unfractionated heparin, enoxaparin, or bivalirudin was administered after insertion of the arterial sheath, and monitoring of the anticoagulation level was recommended according to local laboratory practice (e.g., activated clotting time ≥250 seconds). The use of glycoprotein IIb/IIIa inhibitors was at the discretion of the operator. Initial balloon angioplasty of the target lesion was recommended, though direct stenting was permitted in the study protocol. The PROMUS Element EES was available in diameters of 2.25, 2.5, 2.75, 3.0, 3.5, and 4.0 mm and in lengths of 8, 12, 16, 20, 24, 28, 32, and 38 (>2.25 mm) mm. The XIENCE PRIME EES was available in identical diameters and in lengths of 8, 12, 15, 18, 23, 28, 33 (>2.25 mm), and 38 (>2.25 mm) mm. Treatment of non-target lesions with devices other than the study drug-eluting stent (DES) was not permitted; however, staged procedures were allowed for multiple lesions within 42 days of the index intervention and using the assigned randomised DES.
Within 24 hours prior to the scheduled PCI, all patients had a 12-lead electrocardiogram (ECG), and assessment of creatinine kinase (CK), CK-MB, and cardiac troponin levels. These tests were repeated within 24 hours after the PCI or prior to hospital discharge, whichever occurred first, or in suspected cases of myocardial ischaemia. At hospital discharge, all patients were required to take aspirin 75-160 mg daily indefinitely, and either clopidogrel 75 mg daily or prasugrel 10 mg daily for at least six months.
ENDPOINTS AND DEFINITIONS
The primary endpoint was target vessel failure (TVF) at 12 months following index PCI. TVF was defined as the composite of cardiac death related to the target vessel, MI (Q-wave and non-Q-wave) related to the target vessel, or ischaemia-driven revascularisation of the target vessel (TVR). Cardiac death was defined as death due to: MI; arrhythmia or conduction disturbance; deaths related to the procedure; stroke prior to hospital discharge; and death of unknown cause. Periprocedural MI (≤48 hours) was defined as follows: Q-wave MI: new pathological Q-waves in ≥2 leads lasting ≥0.04 seconds with post-procedure CK-MB >upper limit of normal (ULN) (or troponin >1×ULN with normal baseline); non-Q-wave MI: de novo CK elevation >3.0×ULN (or troponin >3×ULN with normal baseline) in the absence of CK without new Q-waves, and the presence of ECG changes indicative of new ischaemia (new ST-T changes or left bundle branch block [LBBB]), imaging evidence of new loss of viable myocardium, or new regional wall motion abnormality. Spontaneous MI was defined as follows: Q-wave MI: new pathological Q-waves in ≥2 leads lasting ≥0.04 seconds with post-procedure CK-MB >ULN (or troponin >1×ULN with normal baseline); de novo CK elevation >2.0×ULN (or troponin >2×ULN with normal baseline) without new Q-waves, and the presence of ECG changes indicative of new ischaemia (new ST-T changes or LBBB), imaging evidence of new loss of viable myocardium, or new regional wall motion abnormality. TLR was defined as ischaemia-driven repeat PCI to the target lesion or coronary artery bypass grafting (CABG) distal to the target lesion. TLR was considered to be ischaemia-driven if the target lesion diameter stenosis was ≥70% by visual assessment or ≥50% by visual assessment with supporting evidence of clinical or functional ischaemia.
Pre-specified secondary endpoints were adjudicated at 30 days and one year, and included: TVF; ischaemia-driven target lesion revascularisation (TLR); ischaemia-driven target vessel revascularisation (TVR); MI (Q-wave and non-Q-wave); cardiac death; non-cardiac death; all-cause death or MI; all-cause death/MI/TVR; major adverse cardiac events (MACE) rate, defined as a composite of death, MI (Q-wave or non-Q wave), emergent CABG, or TLR; and stent thrombosis according to the ARC definition of definite and probable stent thrombosis and categorised as early, late or very late13. TVR was defined as target vessel-related cardiac death, MI, or ischaemia-driven TVR (TLR or ischaemia-driven revascularisation of a new lesion within the target vessel but excluding the target lesion itself). Non-cardiac death was defined as a death not due to cardiac causes as outlined above. Procedural success was defined as mean lesion diameter stenosis <30% in two near-orthogonal projections with TIMI 3 flow, as visually assessed by the physician, without in-hospital MI, TVR, or cardiac death.
All patient data were collected using an internet-based secure electronic data capture system. Accuracy and completeness of the recorded data were ensured by the site principal investigators and were verified by monitoring visits and evaluation of original source documents by the contract research organisation (CERC, Massy, France). All angiographic data were visually assessed by the enrolling centre. A blinded independent clinical events committee reviewed and adjudicated all clinical endpoints and adverse events, including deaths, suspected MI, TLR, TVR, and stent thrombosis. An independent data monitoring committee, comprised of interventional cardiologists and biostatisticians, reviewed accumulating safety data to monitor the incidence of adjudicated and non-adjudicated events and other trends that could warrant modification or termination of the trial. Members of the data monitoring committee were unaware of stent allocation but had the authority to unblind if required.
STATISTICS
The PLATINUM PLUS trial was designed as a non-inferiority study, powered for non-inferiority of the primary endpoint at 12 months. Given the reported incidence of one-year TVF in the XIENCE V arms of the SPIRIT IV and V trials8,14, we projected a 12-month TVF rate of 7% for both the PROMUS Element and the XIENCE PRIME stents. If the upper limit of the one-sided 95% confidence interval (CI) of the absolute risk difference was <3%, non-inferiority would be declared. Based on these assumptions and a 2:1 randomisation, we estimated that 1,987 patients would be required in the PROMUS Element group and 993 in the XIENCE PRIME group (total: 2,980). These estimates included a 10% expected rate of attrition and yielded 80% power to detect non-inferiority at a one-sided type 1 error of 0.05. We used a two-group Farrington-Manning test to evaluate the one-sided hypothesis of non-inferiority in proportions15. Between-group differences were compared using the chi-square test or Fisher’s exact test for discrete variables and the Student’s t-test for continuous variables. Kaplan-Meier plots of time-to-event variables were constructed and treatment groups were compared using the log-rank test. The primary and additional endpoints were analysed both on an intent-to-treat and on a per-protocol basis. For intent-to-treat analyses, all patients providing informed consent and enrolled in the study were included in the analysis, regardless of whether or not a study stent (PROMUS Element or XIENCE PRIME) was implanted. In the per-protocol analyses, only patients who had the assigned study stent implanted in the target coronary artery were included in the analysis. Other than the one-sided 0.05 test for non-inferiority, all other p-values are two-sided. All statistical analyses were performed using SAS system software, version 8 or above (SAS Institute Inc., Cary, NC, USA).
Results
The study flow is depicted in Figure 1. Between February 2010 and October 2012, a total of 2,980 patients were randomised to receive the PROMUS Element (n=1,952) or XIENCE PRIME (n=1,028) DES. The baseline clinical and angiographic characteristics of the study groups were well matched (Table 1). The mean age was 65.8±10.6 years, 77.9% were male, and 29.4% had diabetes mellitus. One third (36.9%) presented with acute coronary syndromes. The left main was the target vessel in 6.5% and the left anterior descending was the most frequently treated vessel (65.9%). Almost half of all patients (48.3%) had multiple target vessels with an average 1.6±0.9 target lesions per patient.
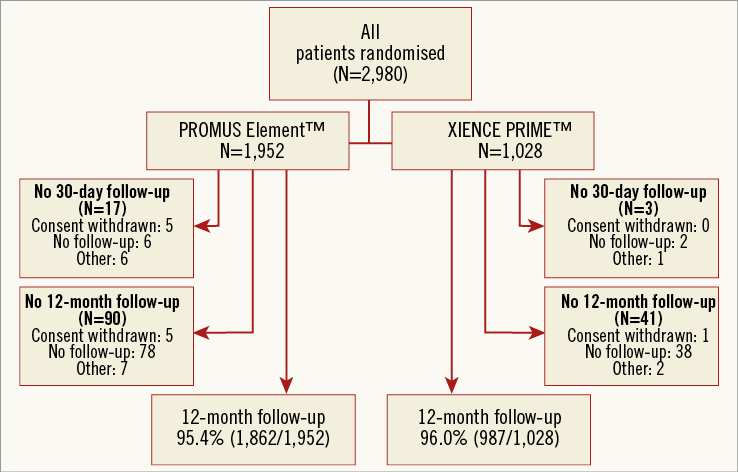
Figure 1. Flow chart and follow-up of the study population.
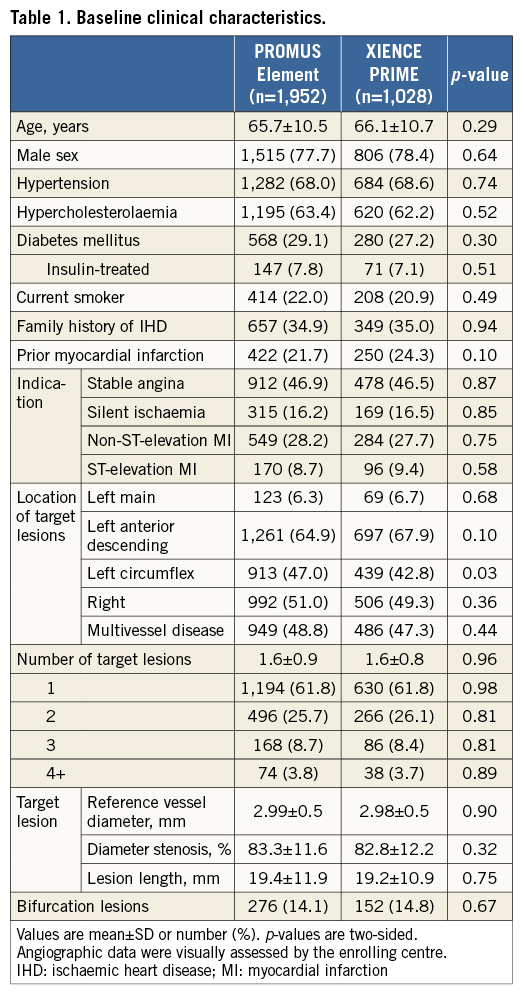
Procedural and angiographic details are presented in Table 2. These characteristics were well matched between groups. The average stent length and diameter per target lesion were 23.93±13.87 mm and 3.00±0.47 mm, respectively. The mean number of stents per target lesion was 1.18±0.49 and, on average, each patient received 1.73±1.11 stents. Procedural success was observed in 97.6% in the PROMUS Element group and in 97.8% in the XIENCE PRIME group (p=0.78). The necessity to implant a second stent due to edge dissection occurred in 49 (2.5%) and 28 (2.7%) patients of the PROMUS Element and XIENCE PRIME groups, respectively (p=0.72).
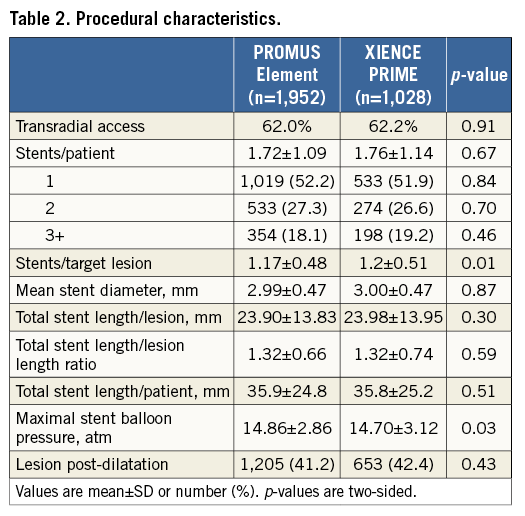
CLINICAL OUTCOMES
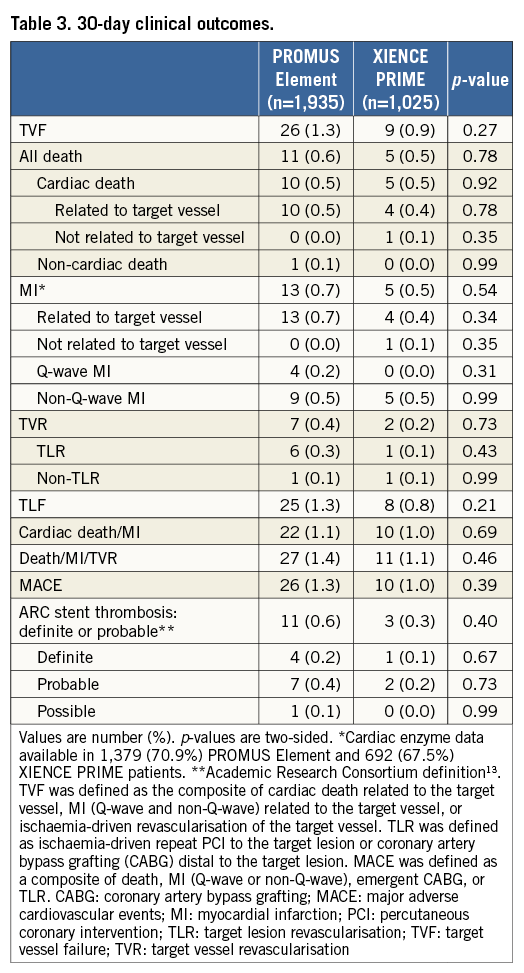
As depicted in Figure 1, 30-day follow-up was available in 99.1% of the PROMUS Element group and in 99.7% of the XIENCE PRIME group (p=0.11). The rates of adverse events were similar in both groups at 30 days (Table 3). In the intention-to-treat analysis, TVF occurred in 1.3% of the PROMUS Element group and in 0.9% of the XIENCE PRIME group (p=0.27).
At 12 months, the primary endpoint occurred in 86 (4.6%) patients with the PROMUS Element stent and in 32 (3.2%) with the XIENCE PRIME stent (Figure 2). Within the pre-specified non-inferiority criteria, the PROMUS Element stent was deemed to be non-inferior to the XIENCE PRIME stent, with an absolute risk difference of 1.4% (95% CI: –0.1-2.9), and the upper limit of the one-sided 95% CI at 2.57%, which was less than the pre-specified non-inferiority margin of <3.0% (one-sided p-value for non-inferiority=0.012) (Figure 3). In the superiority analysis, the platforms were not statistically different (hazard ratio [HR] 1.44, 95% CI: 0.96-2.16, p=0.08).

Figure 2. Kaplan-Meier curves for the primary endpoint at 12 months.

Figure 3. TVF: primary endpoint at 12 months (ITT).
The per-protocol analysis included 1,880 patients in the PROMUS Element group and 973 in the XIENCE PRIME group. The rate of TVF was 4.5% (81/1,799) and 2.8% (26/934) in the PROMUS Element and XIENCE PRIME groups, respectively. The absolute risk difference was 1.7% and the upper limit of the one-sided 95% CI at 2.92% was less than the pre-specified non-inferiority margin of <3.0% (one-sided p-value for non-inferiority=0.039). In the superiority analysis, however, the primary endpoint was significantly increased in the PROMUS Element stent compared to the XIENCE PRIME stent (HR 1.64, 95% CI: 1.05-2.55, p=0.03).
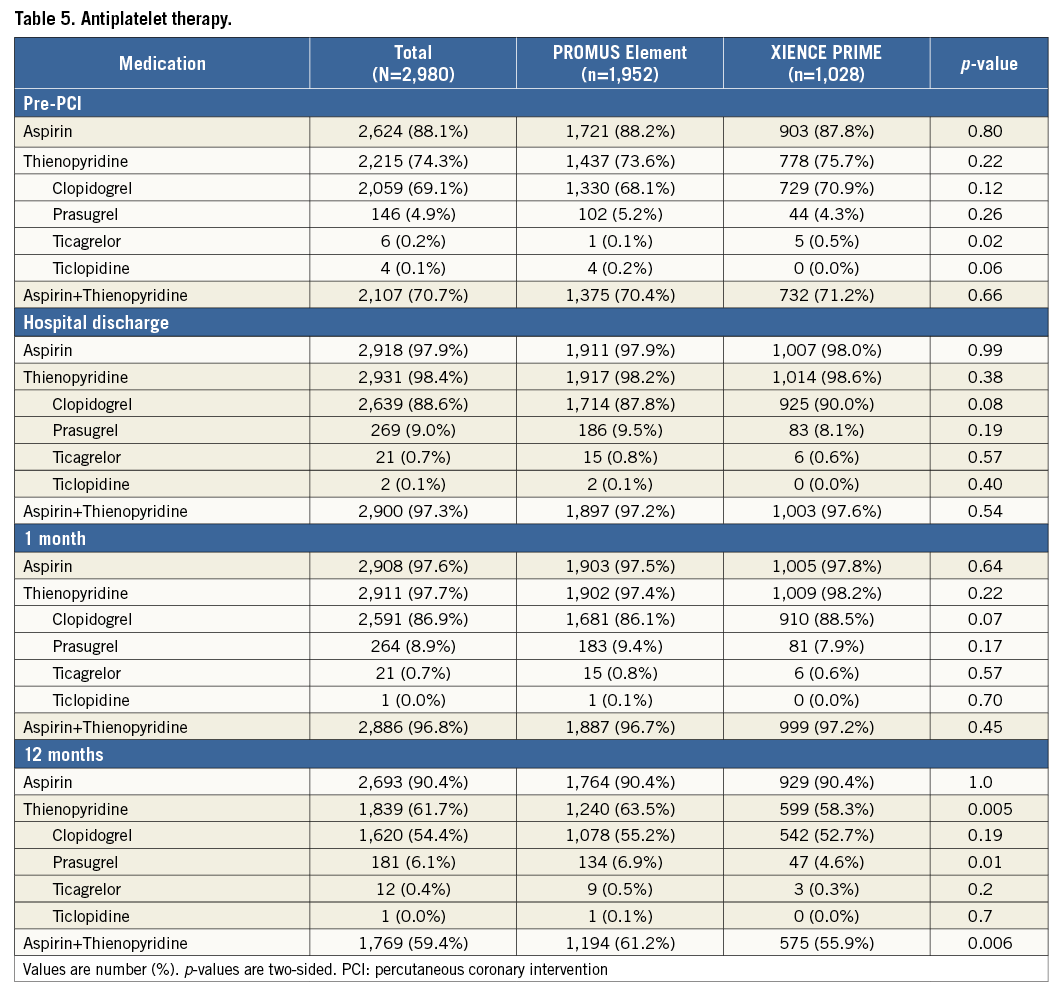
At 12 months, we observed no significant difference in the individual rates of death, cardiac death, MI, and TLR between the PROMUS Element and XIENCE PRIME stents (Table 4, Figure 4). The rates of ARC definite or probable stent thrombosis at 12 months were also similar between groups (0.8% vs. 0.5%, p=0.44). The use of antiplatelet therapies was similar between groups prior to and up to one month after the index intervention; however, dual antiplatelet use was increased in the PROMUS Element group at 12 months (61.2% vs. 55.9%, p=0.006) (Table 5).
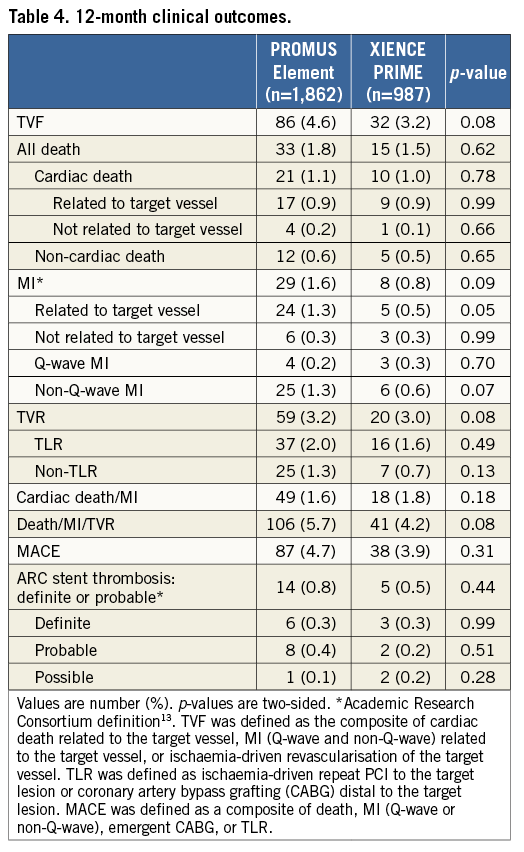
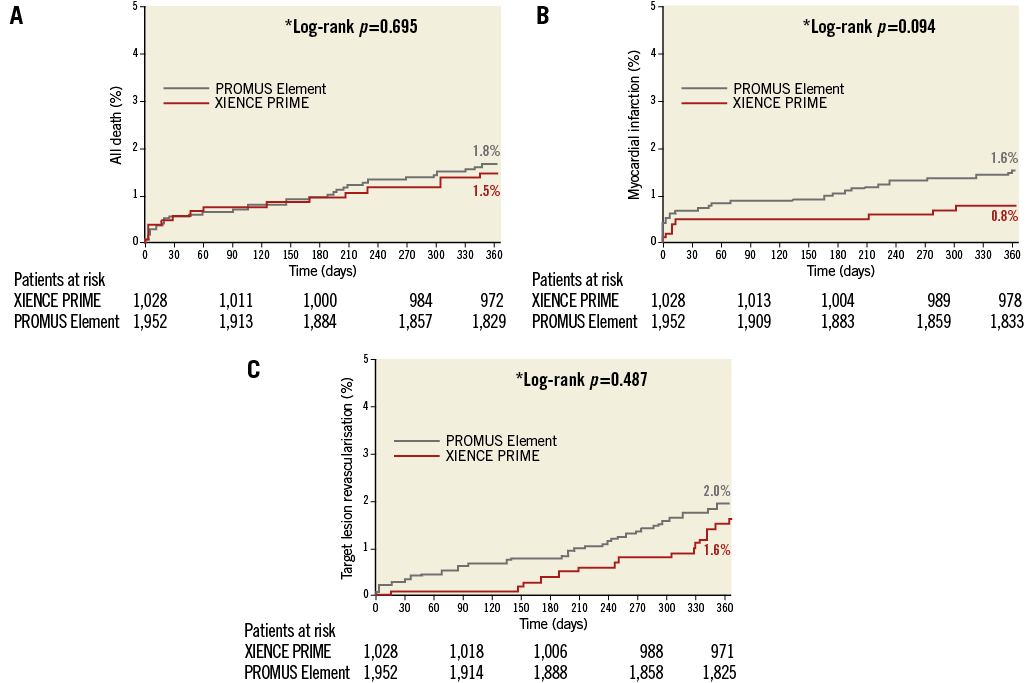
Figure 4. Kaplan-Meier curves for all-cause death, myocardial infarction, and target lesion revascularisation at 12 months (ITT). A) All-cause death. B) Myocardial infarction. C) Target lesion revascularisation.
Pre-specified subgroup analyses included diabetes mellitus, all acute coronary syndromes, STEMI, and de novo lesions (Figure 5). Among these subgroups, no significant between-group differences were observed at 12 months, except in patients with diabetes mellitus, who had an increased rate of TVF with the PROMUS Element stent (7.8% vs. 3.0%, HR 2.64, 95% CI: 1.2-5.83, p=0.01). This result was driven by increased TVR among PROMUS Element recipients (5.1% vs. 2.1%, p=0.06).
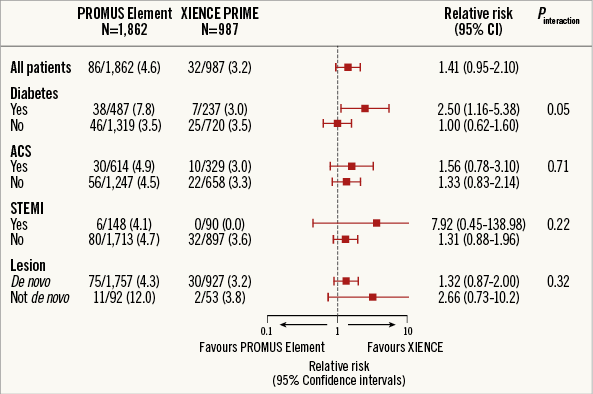
Figure 5. Subgroup analyses (ITT).
Discussion
The current multicentre randomised all-comers PLATINUM PLUS clinical trial demonstrated that the rate of TVF with the PROMUS Element EES was non-inferior to that of the XIENCE PRIME EES at 12-month follow-up, according to the pre-specified non-inferiority criteria. Indeed, this study observed good safety and efficacy for both EES platforms, with low rates of MI, stent thrombosis and ischaemia-driven TLR in a real-world patient population.
Large randomised trials have previously observed superior performance with cobalt-chromium EES compared to PES1,3,16. The supremacy of the new-generation cobalt-chromium EES was manifest in lower rates of TVF, TLR, MI, and stent thrombosis than the first-generation PES. Recently published data also suggest a potential mortality advantage of cobalt-chromium EES over PES17. Cobalt-chromium EES have also demonstrated similar performance to second-generation ZES (Resolute™; Medtronic CardioVascular, Santa Rosa, CA, USA)18-20.
The continued development of DES has focused on enhancing each of the three major elements of the stent platform: the stent scaffold, the polymer, and the antiproliferative agent. The PROMUS Element DES comprises a novel platinum-chromium stent with modified stent architecture, and the previously tested durable biocompatible polymer and antiproliferative agent8. Specifically, the design goals of the platinum-chromium scaffold were to improve deliverability, radial strength, vessel conformability, radiopacity, and side branch accessibility, without compromising the safety and efficacy profile of the stent4,5. In this regard, animal models have suggested that the platinum-chromium EES yields drug-elution kinetics and drug tissue concentrations similar to its cobalt-chromium equivalent21.
Supporting clinical data come from the PLATINUM trial8. This prospective study randomised 1,530 patients undergoing PCI for de novo stable coronary artery disease to treatment with the PROMUS Element or XIENCE V EES. At one year, the platinum-chromium EES was non-inferior to its cobalt-chromium equivalent (TVF 3.4% vs. 2.9%; absolute risk difference 0.5%, 95% CI: –1.3-2.3; upper limit of one-sided 95% CI: 2.13%; non-inferiority p=0.001). While these data suggest clinical equipoise between the EES platforms, patients with acute MI, true bifurcation lesions, left main stenosis, and chronic total occlusions were excluded from the PLATINUM study. Consequently, this study does not provide insight into the majority of patients undergoing PCI in a real-world setting10,11. As such, the Circulatory System Devices Advisory panel of the FDA recommends that all new DES be tested in well-designed trials in off-label indications12. The recently reported phenomenon of longitudinal stent deformation underscores the need to undertake all-comer comparative safety and effectiveness studies22. While the all-comers trial design is considered to be reasonably representative of “real-world” clinical practice, this design continues to exclude some patient groups, and in particular higher-risk patients9.
The current randomised trial included the largest number of patients (N=2,980) randomised to platinum-chromium or cobalt-chromium EES. Over one third of patients presented with acute MI and almost half had multivessel disease. Although the proportion of patients with acute coronary syndromes (36.9%) was lower than that in previous all-comer designs such as the DUTCH PEERS (TWENTE II) study, the proportion of patients undergoing left main (6.5%) or multivessel PCI (48.3%) was higher, thereby suggesting a legitimately high-risk study population9. In this all-comer population, the PROMUS Element EES satisfied the non-inferiority criteria relative to the XIENCE PRIME EES for the composite safety and efficacy endpoint of TVF at 12 months. In contrast, the per-protocol analysis suggested that the control XIENCE PRIME EES might be superior to the PROMUS Element EES (HR 1.6; p=0.03). The reasons for such divergent results are unclear. Longitudinal stent deformation (LSD), which has been recognised as a potential cause of stent failure22,23, has in some analyses been more frequently associated with the PROMUS Element platform24,25. Although various other studies have suggested that the PROMUS Element platform has a similar propensity towards LSD26-29, the PROMUS Element stent has recently been redesigned to include, amongst other modifications, additional connectors at the proximal end to reduce the risk of deformation (Synergy systems)30. The aforementioned discrepant result could also be attributed to other differences between the stent platforms, or simply to the play of chance.
Acute procedural success (>97%) and the requirement for implantation of a second stent due to edge dissection was similar between study platforms. The requirement for a higher rate of “bail-out” stenting with the XIENCE PRIME stent in the PLATINUM study was not observed in the current study8. Nevertheless, there were no significant differences in the individual rates of death, MI, or ischaemia-driven TLR between the groups at one year. Moreover, the one-year incidence of ARC-defined definite or probable stent thrombosis was reassuringly low in both the platinum-chromium (0.8%) and cobalt-chromium (0.5%) EES. Indeed, the one-year rates of TVF with the PROMUS Element (4.6%) and XIENCE PRIME (3.2%) EES platforms were considerably lower than the expected rate (7%) for each stent. Nevertheless, in the non-inferiority analysis, this difference remained within the pre-specified 3% margin, despite a weak, non-significant trend (p=0.08) towards increased TVF at 12 months with the PROMUS Element EES. This trend was largely driven by a numerical increase in non-Q-wave MI (1.3% vs. 0.6%, p=0.07), TLR (2.0% vs. 1.6%, p=0.49), and non-target lesion TVR (1.3% vs. 0.7%, p=0.13) with the PROMUS Element stent compared to the XIENCE PRIME stent. Furthermore, subgroup analysis demonstrated an increased rate of TVF in PROMUS Element-treated patients. The clinical relevance of this finding is uncertain, as the current study was not designed to evaluate differences in outcome in patients with diabetes. These data underscore the need for longer-term follow-up or perhaps larger randomised trials, specifically tailored to patients with diabetes mellitus, to demonstrate clinical equipoise between these platforms unequivocally.
Recently, the PROMUS Element EES was compared to the cobalt-chromium-based ZES in an investigator-initiated prospective single-blind all-comer randomised trial9. Among 1,811 patients, 45% presented with acute coronary syndromes. The primary outcome measure (TVF) occurred in 5% of platinum-chromium EES-treated and 6% of the ZES-treated patients (absolute risk difference 0.88%, 95% CI: –1.24-3.01%; upper limit of one-sided 95% CI: 2.69%; non-inferiority p=0.006). These data, when considered together with the current study, would appear to confirm that the platinum-chromium EES provides similar efficacy and safety to third-generation cobalt-chromium EES and ZES.
Limitations
This study has limitations. The proportion of patients in each participating institution who were randomised to the trial protocol during the enrolment period was not recorded, thus precluding an objective appraisal of the all-comers enrolment process. The proportion of STEMI patients undergoing primary PCI within 24 hours was not recorded, nor was the documentation of ischaemia in patients undergoing PCI for silent ischaemia. The rate of periprocedural MI in the current study was low (30-day MI: 0.7%), and is most likely due to underreporting: post-PCI CK/CK-MB and/or troponin data were only available in 1,379 (70.9%) PROMUS Element and 692 (67.5%) XIENCE PRIME patients. At 12-month follow-up, the rate of TVF in the XIENCE PRIME control arm was 3.2%, and was consequently less than the expected 7% rate used for the sample size calculation. Thus, although the absolute difference in TVF at 12 months was small (1.4%), the current study cannot absolutely exclude small differences in event rates between the investigated stents. As in all studies, extended follow-up is required to demonstrate equivalent longer-term outcomes. A dedicated core laboratory quantitative coronary analysis was not performed, thus precluding the analysis of longitudinal stent deformation.
Conclusions
In this large-scale all-comers prospective randomised trial, the platinum-chromium EES was found to be non-inferior to the control cobalt-chromium EES at one-year follow-up.
| Impact on daily practice Based on one-year follow-up, the findings of this large randomised trial in which 3,000 patients were included demonstrate that PCI treatment using platinum-chromium everolimus-eluting stents (PROMUS Element) can be implemented as efficiently and safely as with cobalt-chromium everolimus-eluting stents (XIENCE PRIME). |
Conflict of interest statement
J. Fajadet receives educational grants from Abbott Vascular, Boston Scientific, Medtronic & Terumo. M. Spence is a consultant to Boston Scientific and proctor for the Boston Scientific Lotus™ transcatheter aortic valve. A. Zaman has received honoraria for lectures and attendance at advisory boards from Boston Scientific, Abbott Vascular and Medtronic. S. Elhadad is a consultant to Boston Scientific. F-J. Neuman’s institution has received research grants and speaker honoraria from Boston Scientific and Abbott Vascular. The other authors have no conflicts of interest to declare.
