- coronary artery disease
- intravascular ultrasound
- PROMUS Element
- quantitative coronary angiography
- restenosis
Abstract
Aims: Assess clinical, angiographic, and intravascular ultrasound results in lesions treated with the PROMUS Element platinum chromium everolimus-eluting stent (EES).
Methods and results: Patients (N=100) with one de novo target lesion ≤34mm long and reference vessel diameter (RVD) ≥2.25−≤4.25 mm were enrolled at 14 sites. The primary endpoint was the 30-day composite of cardiac death, myocardial infarction, target lesion revascularisation (TLR), or definite/probable stent thrombosis (ST). The efficacy endpoint of 9-month in-stent late loss in workhorse lesions (defined as RVD ≥2.5−≤4.25 mm, lesion ≤24mm) was compared to a performance goal based on historical results with TAXUS Express paclitaxel-eluting stents. Post-procedure incomplete stent apposition (ISA) was compared to a performance goal based on results with the PROMUS/XIENCEV EES in SPIRITIII. Mean age was 61.8±9.9 years; 77.0% were male; 19% had medically treated diabetes. Baseline RVD was 2.72±0.53mm; lesion length was 15.4±7.0mm. The primary endpoint occurred in one patient (periprocedural ST with TLR) with no additional major clinical events through one year. Nine-month in-stent late loss in workhorse lesions (0.17±0.25 mm, N=73) and post-procedure ISA (5.7%, 5/88) were below performance goals (p<0.001).
Conclusions: Through one year, PROMUS Element EES had an acceptable safety and efficacy profile with low in-stent late loss and post-procedure ISA.
Introduction
While a clear restenosis advantage of coronary drug-eluting stents (DES) over bare-metal stents (BMS) has been demonstrated in randomised, controlled trials (RCT), about 7-10% of patients treated with DES still require repeat revascularisation1-4. Furthermore, first generation DES have shown higher rates of very late stent thrombosis (ST) compared with BMS5. PROMUS® (XIENCEV®), a second-generation everolimus-eluting stent (EES), has shown reduced angiographic late loss, target lesion revascularisation (TLR), myocardial infarction (MI), and ST compared to paclitaxel-eluting stents6-9.
Despite these improvements in clinical outcomes, there is still a need for better performing stents. In the PROMUS Element™ EES, the same drug and polymer coating used in PROMUS is applied to a novel platinum chromium stent platform designed to improve deliverability, radiopacity, radial strength and recoil 10,11. In a rabbit endothelial cell denudation model, the thinner strut bare-metal Element stent exhibited more rapid strut coverage and endothelialisation than Liberté or Express12, and in a non-injured porcine coronary revascularisation model, PROMUS Element demonstrated vascular compatibility equivalent to PROMUS (XIENCEV)13. On the basis of these promising preclinical data, PLATINUM QCA (Prospective, Randomised, MuLticenter TriAl To Assess an EverolImus-Eluting CoroNary Stent System [PROMUS EleMent] Quantitative Coronary Angiography), a prospective, single-arm, multicentre observational study, was designed to evaluate the clinical, angiographic and intravascular ultrasound (IVUS) outcomes in lesions treated with the PROMUS Element stent.
Methods
Device description
The PROMUS Element EES (Boston Scientific Corporation, Natick, MA, USA [BSC]) has been described previously11. In brief, the thin-strut, balloon-expandable stent consists of a novel platinum chromium alloy10 with the antiproliferative agent everolimus14 (100μg/cm2) applied in a durable, biocompatible acrylic polymer and fluorinated copolymer15 identical to that used in the PROMUS (XIENCEV) EES3 (manufactured and distributed by Abbott Vascular, Santa Clara, CA, USA, as XIENCEV and distributed by BSC as PROMUS). PROMUS Element and PROMUS (XIENCEV) are shown in Figure1.

Figure 1. PROMUS Element above and PROMUS (XIENCEV) everolimus-eluting stents.
Study design and procedure
Enrolled patients presented with stable or unstable angina pectoris or documented silent ischaemia and were to be treated with one stent for a singlede novo target lesion ≤34 mm long with a visually estimated diameter stenosis ≥50% to <100% in an artery with a reference vessel diameter (RVD) of ≥2.25mm to ≤4.25 mm. Treatment of one non-target lesion in a non-target vessel with a commercial treatment (e.g., stent, balloon angioplasty, excluding brachytherapy) was also allowed provided it occurred before target lesion intervention and was a clinical and angiographic success (defined as visually assessed mean lesion diameter stenosis <50% [<30% for stents] with TIMI 3 flow without prolonged chest pain or MI). Staged or planned revascularisations following the index procedure were prohibited. Patients with myocardial infarction within 72 hours prior to the index procedure and elevated cardiac markers at the time of the procedure were excluded from enrolment. Complete inclusion and exclusion criteria are provided in Appendix A. Clinical follow-up was scheduled at 1, 9, and 12months along with 9-month angiographic and IVUS follow-up. Dual antiplatelet therapy (aspirin and a thienopyridine) was mandated to reduce the risk of thrombosis. Thienopyridine treatment (clopidogrel, ticlopidine, or prasugrel in accordance with approved country-specific labelling) was required for at least sixmonths (12months or longer recommended); aspirin was required indefinitely.
The Ethics Committee at each participating centre approved the study protocol. Informed written consent prior to enrolment or the performance of any study-specific procedures or tests was required from all patients. An independent clinical events committee (CEC) adjudicated all reported events of death, MI, target vessel revascularisation (TVR) and ST, and an independent data monitoring committee provided oversight of aggregate safety data. Angiograms were evaluated by an independent angiographic core laboratory (Beth Israel Deaconess Medical Center, Boston, MA, USA) using software from Medis Medical Imaging Systems (Leiden, The Netherlands). Intravascular ultrasound data were evaluated at an independent core laboratory (MedStar Research Institute, Washington, D.C., USA). Additional PLATINUM study organisation and oversight committee membership are provided in AppendixB. The study is registered at www.clinicaltrials.gov, identifier NCT00824434.
Endpoints
The primary endpoint was the 30-day composite rate of cardiac death, MI, TLR, or ST defined as definite or probable per the Academic Research Consortium [ARC] definitions18. Stent thrombosis was included in the primary endpoint given the concern about ST in the interventional community. Additional endpoints included technical and clinical procedural success (defined in AppendixC); death, MI, revascularisation, and ST at each follow-up period; and QCA and IVUS analyses at ninemonths. The efficacy endpoints, 9-month in-stent late loss by QCA in workhorse lesions (defined as RVD ≥2.5 mm and ≤4.25 mm and lesion length ≤24mm by visual estimate) and post-procedure incomplete stent apposition (ISA) by IVUS, were compared to pre-specified performance goals (see “Statistical methods”). Incomplete stent apposition was defined as separation of one or more stent struts from the vessel wall with evidence of blood speckles behind the stent strut on IVUS. Additional endpoint definitions are provided in AppendixC.
Statistical methods
The efficacy endpoints of 9-month in-stent late loss (in workhorse lesions) and post-procedure ISA were compared to predefined performance goals (PG). For 9-month in-stent late loss, the PG of 0.44mm (0.41mm + delta [0.03mm]) was based on historical results in similar lesions treated with the TAXUS Express stent in the TAXUS IV and TAXUSV studies and the data were compared using a one group t-test (p<0.05 for significance). Assuming an expected in-stent late loss of 0.18±0.50 mm and a sample of 60 workhorse patients, the study had 99% power to conclude in-stent late loss was less than the PG. This conclusion would be made if the one-sided upper 95% confidence bound on the observed value was below the PG. For post-procedure ISA, the PG (34.4%) was based on historical PROMUS (XIENCEV) post-procedure ISA data from the SPIRIT III study,7 and the data were compared using a one-group exact test (p<0.05 for significance). Assuming an expected ISA rate of 17.2% and a sample of 70patients, the study had 95% power to conclude the ISA rate was below the PG if the one-sided Clopper-Pearson exact upper 95% confidence bound on the observed value was less than the PG. No statistical testing was done for the additional endpoints. Patient, lesion, and procedural characteristics and event rates were analysed using descriptive statistics; simple proportions with 95% confidence intervals were used for categorical variables with continuous data provided as mean ± standard deviation. All statistical analyses were performed using SAS version 8 or higher (SAS Institute, Inc., Cary, NC, USA).
Results
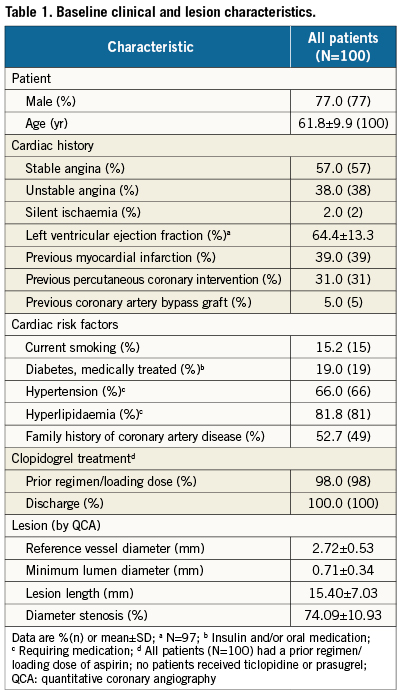
Patient, lesion, and procedural characteristics
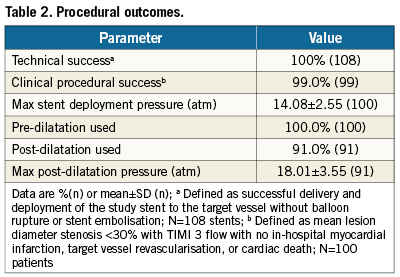
The PLATINUM QCA study enrolled 100patients at 14sites in Australia, Malaysia, New Zealand, and Singapore (AppendixD) from March to July 2009. Baseline demographics and lesion characteristics are shown in Table1. The cohort included 85 workhorse, 12 long-lesion (RVD≥2.25-≤4.25mm, lesion >24-≤34mm), and three small-vessel (RVD≥2.25-<2.5mm, lesion ≤28mm) target lesions (by visual estimate). Procedural outcomes appear in Table2. Device technical success, defined as successful delivery and deployment of the study stent to the target lesion without balloon rupture or embolisation was 100%.
Clinical outcomes at 30 days and one year
Clinical follow-up was 100% at 30days and one year; outcomes are shown in Table3. The primary endpoint, the 30-day composite rate of cardiac death, MI, TLR or ST, occurred in one patient (1.0%) who had a periprocedural ST with TLR and a non-target lesion TVR. There were no additional major clinical events through oneyear. The single patient with an event was a 62-year-old female with a previous triple bypass graft who received a 3.5x12 mm stent implanted in the proximal left anterior descending artery (LAD). One dayafter the index procedure the patient had chest pain and anterior ST-segment elevation which improved after treatment with heparin, nitroglycerine, and tirofiban. Angiography showed a widely patent stent, but there was a slight haziness suggestive of thrombus at the origin of an adjacent branch vessel. There was also severe vessel spasm in the distal LAD. Assessment by IVUS showed stent malapposition and further balloon angioplasty of the stent was performed. The maximum CK and CK-MB after the event were 328 U/L (upper limit of normal [ULN] 190 U/L) and 13U/L (ULN 10 U/L).

Quantitative coronary angiography
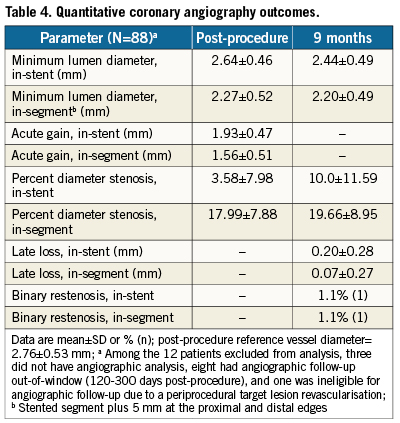
Follow-up at 9-months was 88% for QCA. Nine-month in-stent late loss in workhorse lesions was 0.17±0.25 mm (N=73) with an upper confidence bound of 0.22 mm, significantly below the pre-specified performance goal (p<0.001). Figure2 shows cumulative frequency distribution curves for in-stent percent diameter stenosis (DS) and in-stent minimum lumen diameter (MLD) in workhorse lesions pre-procedure, post-procedure, and at follow-up. The mean pre-procedure DS of 72.95±10.63% was reduced to 3.57±8.32% after stent implantation and remained low at 9.34±10.44% by ninemonths. Mean MLD was 0.75±0.34mm before stent implantation, 2.68±0.44mm post-procedure, and 2.50±0.46mm at follow-up. Table4 shows QCA results in all lesion types (N=88). At ninemonths, in-stent late loss was 0.20±0.28mm and binary restenosis was 1.1% (N=1).
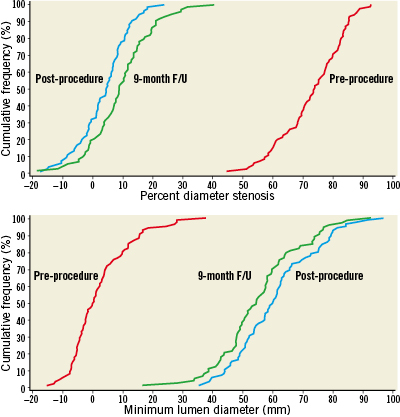
Figure2. Cumulative frequency distribution curves in workhorse lesions; red: pre-procedure, blue: post-procedure, green: 9-month follow-up. In-stent percent diameter stenosis (above); In-stent minimum lumen diameter.
Intravascular ultrasound
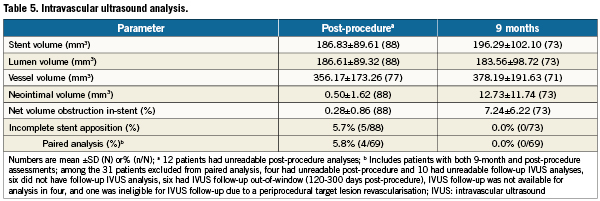
Post-procedure ISA was present in 5.7% of patients (5/88, upper confidence bound of 11.6%), significantly less than the predetermined performance goal of 34.4% reported for PROMUS (XIENCE V) in the SPIRIT III trial (p<0.001)7. Follow-up at 9-months was 83% for IVUS. Among patients with paired analyses (N=69), all post-procedure ISA was resolved at ninemonths with no late acquired ISA. Percent volume obstruction in 73 lesions was 7.2±6.2 at ninemonths. Additional IVUS outcomes are shown in Table5.
Discussion
This prospective multicentre study evaluated the clinical, angiographic, and intravascular ultrasound outcomes in de novo coronary stenoses treated with the thin-strut, platinum chromium PROMUS Element everolimus-eluting stent. The primary endpoint (30-day composite of cardiac death, MI, TLR, or ST) occurred in 1.0% of patients (1/100, with one patient having a periprocedural ST with TLR). There were no additional major cardiac events through one year. The performance goals for the angiographic and IVUS efficacy measures were met as in-stent late loss in workhorse lesions (RVD ≥2.5–≤4.25mm, lesion length ≤24mm; N=73) was 0.17±0.25mm compared to a value of 0.44mm based on historical TAXUS Express results (P<0.001)2,19 and post-procedure ISA (N=88) was 5.7% versus 34.4% for PROMUS (XIENCE V) in SPIRIT III (p<0.001)7. These outcomes suggest that the everolimus/polymer combination in PROMUS (XIENCEV) can be successfully transferred to the platinum chromium Element stent platform.
The mean in-stent late loss of 0.17 mm at nine months with PROMUS Element in workhorse lesions is comparable to that previously reported for PROMUS (XIENCE V) in the SPIRIT First trial (0.10±0.21mm at sixmonths3 and 0.24±0.27mm at oneyear20), SPIRITII (0.11±0.27mm at sixmonths6), and SPIRITIII (0.16± 0.41mm at eightmonths7). Late loss in-segment at ninemonths with PROMUS Element in all lesions was 0.07±0.27mm (N=88). This result was also similar to that reported for in-segment late loss with PROMUS (XIENCE V) in SPIRIT First (0.07±0.19mm [sixmonths]3; 0.14±0.24mm [one year]20), SPIRITII (0.07±0.33mm [six months]6), and SPIRIT III (0.14±0.41mm [eight months]7).
Intravascular ultrasound outcomes at nine months in PLATINUM QCA were similar to outcomes reported for PROMUS (XIENCEV) in the SPIRIT trials. Neointimal hyperplasia was 12.7±11.7mm3 and percent volume obstruction was 7.2±6.2 (N=73) with PROMUS Element compared to 10.1±11.5mm3 and 6.9±6.4% (N=101), respectively, for PROMUS (XIENCE V) at eightmonths in SPIRITIII7. Similar outcomes were reported at sixmonths in SPIRIT First (10±13mm3 and 8.0±10.4%, N=21)3 and SPIRIT II (4±7mm3 and 2.5±4.7%, N=100)6.
Post-procedure ISA with PROMUS Element (5.7%, 5/88) was significantly less than that reported for PROMUS (XIENCE V) in SPIRITIII (34.4%, p<0.001). This may reflect differences in stent design, but could also be related to other factors including the extensive use (91.0%) of post-dilatation and/or the maximum post-dilatation balloon pressure (18.0±3.6atm) in the PLATINUM QCA study. Among SPIRIT III patients with both post-procedure and 8-month IVUS follow-up (110 patients, 117 lesions) the post-dilatation rate was 48.7% with 15.7±3.3atm maximum post-dilatation balloon pressure. In this patient subset, post-procedure ISA was 33.3%, which was similar to that observed in the overall SPIRIT III population but significantly greater than the ISA rate observed in the SPIRIT III Japan registry (15.9%, p=0.006; 73 patients with 82 lesions).21 Post-dilatation rates and maximum post-dilatation balloon pressure were higher in the SPIRIT III Japan registry (65.9% and 17.9±2.7atm, respectively).21
Importantly, among patients with paired IVUS analyses there were no occurrences of late acquired ISA with PROMUS Element (0/69, Table5). Indeed, late ISA was not present in any patient treated with PROMUS Element (0/73). The absence of late acquired ISA is consistent with that observed for PROMUS (XIENCE V) (1.1% at eightmonths in SPIRIT III7, none at sixmonths in SPIRIT II6, none at sixmonths or oneyear in SPIRIT First20). Also of note, there was no significant change in vessel dimensions and volume in the PROMUS Element stented segment indicating the absence of positive vessel remodelling due to chronic inflammation.
Study limitations
PLATINUM QCA was a small, first human use, non-randomised study with comparisons to performance goals based on historical results, and there is a potential for bias due to differences in patient complexity or treatment patterns. Operators may also have modified their treatment strategy based on their knowledge of the previously observed rates of post-procedure incomplete apposition. The study was not powered to investigate clinical endpoints; in the PLATINUM Workhorse RCT, a companion trial in the PLATINUM Clinical Trial Program with 1,530 enrolled patients, PROMUS Element was found non-inferior to PROMUS for 12-month target lesion failure and the two stents had similar, low 12-month rates of cardiac death, MI, TLR, and ST22. Finally, results obtained in PLATINUM QCA may not apply to patient and lesion types excluded from enrolment.
Conclusions
Through one year the PROMUS Element everolimus-eluting stent has an acceptable safety and efficacy profile with low in-stent late loss and post-procedure incomplete stent apposition. These results suggest that the drug and polymer combination used in PROMUS (XIENCEV) can successfully be transferred to the platinum chromium Element stent platform.
Acknowledgements
The authors thank Edmund McMullen, M.Math and Peggy Pereda, MS (Boston Scientific Corporation) for assistance with statistical analyses. PLATINUM QCA is sponsored and funded by Boston Scientific Corporation, Natick, MA, USA.
Funding source
Boston Scientific Corporation, Natick, MA, USA
Conflict of interest statement
Prof. Meredith has served as a consultant for BSC, Abbott Vascular, and Medtronic. Dr. Stone is on the Scientific Advisory Board for, and has received honoraria from BSC and Abbott Vascular. Dr.Teirstein reports receiving research funding, consulting fees and honorarium from BSC, Medtronic, Cordis, and Abbott. Drs. Allocco, Dawkins, and Starzyk are full-time employees and stockholders of BSC. The other authors have no conflicts of interest to declare.
Online data supplement
AppendixA. Patient and angiographic inclusion and exclusion criteria.
AppendixB. PLATINUM QCA study organisation and processes.
AppendixC. Definitions.
AppendixD. PLATINUM QCA investigative sites.
