Abstract
Aims: The PROMUS™ Element™ European Post-Approval Surveillance Study (PE-Prove) is a prospective, open-label, multicentre observational study designed to assess outcomes following PROMUS Element everolimus-eluting stent implantation in an unselected patient population.
Methods and results: A total of 1,010 patients were enrolled at 40 clinical sites in Europe, including 24.9% with medically treated diabetes, 50.0% with Type B2/C lesions, 6.1% with chronic total occlusion, 17.8% with acute myocardial infarction (MI ≤24 hours pre-procedure), and 20.1% with unstable angina. The target lesion was the culprit for ST-segment elevation MI in 7.3% of patients. The one-year, per patient target vessel failure rate was 6.2% (60/975), 3.4% (33) being related to the PROMUS Element stent. Rates of cardiac death, MI, and Academic Research Consortium (ARC) definite/probable stent thrombosis were 1.7%, 3.5%, and 0.6%, respectively. The target vessel revascularisation rate was 3.2% (31/975), 2.1% (20) being related to the PROMUS Element stent.
Conclusions: In a large and relatively complex group of “real-world” patients, coronary artery revascularisation with the PROMUS Element everolimus-eluting stent provides favourable results with low event rates consistent with those reported for other contemporary drug-eluting stents.
Introduction
As new coronary stent designs become approved and more widely available for use in large, unselected populations outside of controlled clinical trials, it is important to evaluate their safety and effectiveness in broader “real-world” application. The PROMUS Element™ coronary stent (Boston Scientific Corporation, Marlborough, MA, USA) is a thin-strut, platinum chromium alloy stent coated with a durable, biocompatible, inert fluorocopolymer and everolimus as the antiproliferative agent. In the randomised controlled PLATINUM trial, the PROMUS Element stent was shown to be non-inferior to the predicate XIENCE V/PROMUS stent (Boston Scientific) for the primary endpoint of one-year target lesion failure1. Rates of all-cause death, cardiac death, myocardial infarction, stent thrombosis, and revascularisation were comparable between the two treatment groups at three-year follow-up2.
The PROMUS™ Element™ European Post-Approval Surveillance Study (PE-Prove) is an observational study designed to collect data on long-term outcomes in a large and relatively complex group of “real-world” patients treated with the PROMUS stent. We report here the one-year primary endpoint clinical outcomes, evaluating the safety and efficacy of this stent in a large, unselected patient population.
Methods
This prospective, open-label, multicentre study with an all-comers approach was designed to enrol approximately 1,000 patients at 40 sites in Europe.
PATIENT SELECTION, PROCEDURE, AND FOLLOW-UP
All patients who were candidates for coronary artery stenting and eligible to receive a PROMUS Element stent were evaluated for enrolment in this study. All enrolled patients signed a written informed consent form that had been approved by the independent ethics committee at each study site. Enrolment was considered complete upon signing the informed consent form. This study was conducted in accordance with the ethical principles originating in the Declaration of Helsinki and consistent with good clinical practice and applicable local regulatory requirements (ClinicalTrials.gov NCT01148329).
Follow-up assessments, including medications, NYHA/CCS classification, adverse events, and coronary angiograms performed according to standard of care at each study site, were carried out by clinic visit or phone call at 30 days, six months, and 12 months post index stent implantation, and will continue annually to five years.
STUDY ENDPOINTS
The primary endpoint of this study was the overall and PROMUS Element stent-related target vessel failure (TVF) rate, defined as cardiac death, myocardial infarction (MI) related to the target vessel, or target vessel reintervention (TVR), at 12 months post stent implantation. Secondary endpoints are detailed in the Online Appendix. An independent clinical events committee adjudicated all deaths, MI, TVR, and stent thrombosis, including the relationship of the event to the study stent.
STATISTICAL METHODS
Enrolled patients who received at least one PROMUS Element stent in the target lesion were included in the analysis. Statistical analyses were performed using SAS®, Version 9 or later (SAS Institute Inc., Cary, NC, USA). Further details are provided in the Online Appendix.
Results
PATIENT, LESION, AND PROCEDURAL CHARACTERISTICS
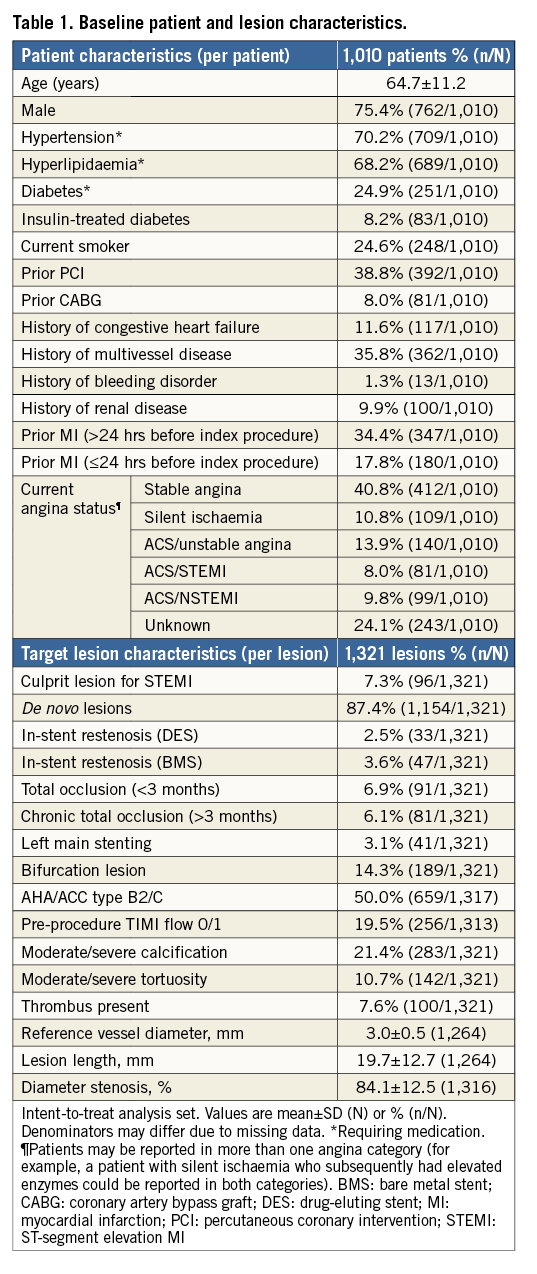
Of 1,010 patients enrolled at 40 clinical sites in Europe between 28 June 2010 and 20 April 2011, one-year clinical follow-up was available for 975 (96.5%) patients (Figure 1). Baseline patient and lesion characteristics are shown in Table 1.

Figure 1. PE-Prove registry enrolment and follow-up.
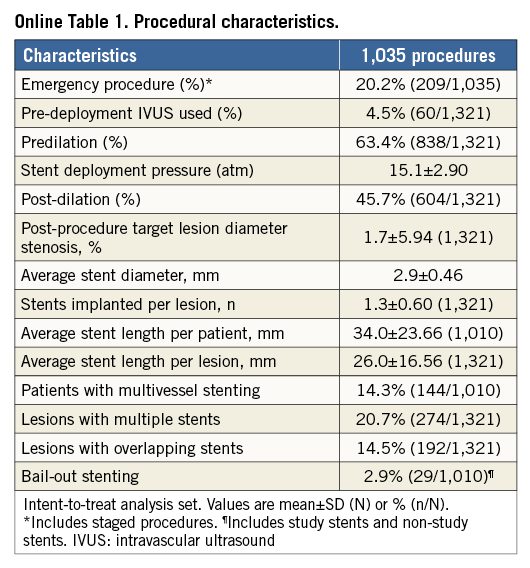
Procedural characteristics are shown in Online Table 1. Successful deployment of the PROMUS Element stent to the target lesion without device malfunction (i.e., technical success) was achieved in 99.8% (1,008/1,010) of patients. Two cases of longitudinal stent deformation were detected by angiography during the index procedure, both of which involved <5% proximal stent compression. Details of these cases are provided in the Online Appendix.
ONE-YEAR CLINICAL OUTCOMES
As shown in Table 2, the overall one-year TVF rate was 6.2% and TVF related to the PROMUS Element stent was 3.4%. The rates of cardiac death, MI, and TVR related to the PROMUS Element stent were 0.4%, 2.1%, and 2.1%, respectively. ARC-defined definite/probable stent thrombosis related to the PROMUS Element stent was reported in six patients (0.6%). In Figure 2, showing Kaplan-Meier time-to-event analyses for overall TVF and definite/probable stent thrombosis at one-year follow-up, there were no stent thromboses reported beyond eight months post procedure. One-year clinical outcomes for selected high-risk patient subgroups are also shown in Table 2. Relative to the study population as a whole, rates of overall and PROMUS Element-related TVF and ARC-defined definite/probable stent thrombosis were only modestly higher in patients with medically treated diabetes or long lesions (>28 mm single lesion), whereas rates among patients with small vessels (≤2.5 mm) were similar to the overall study population rates.
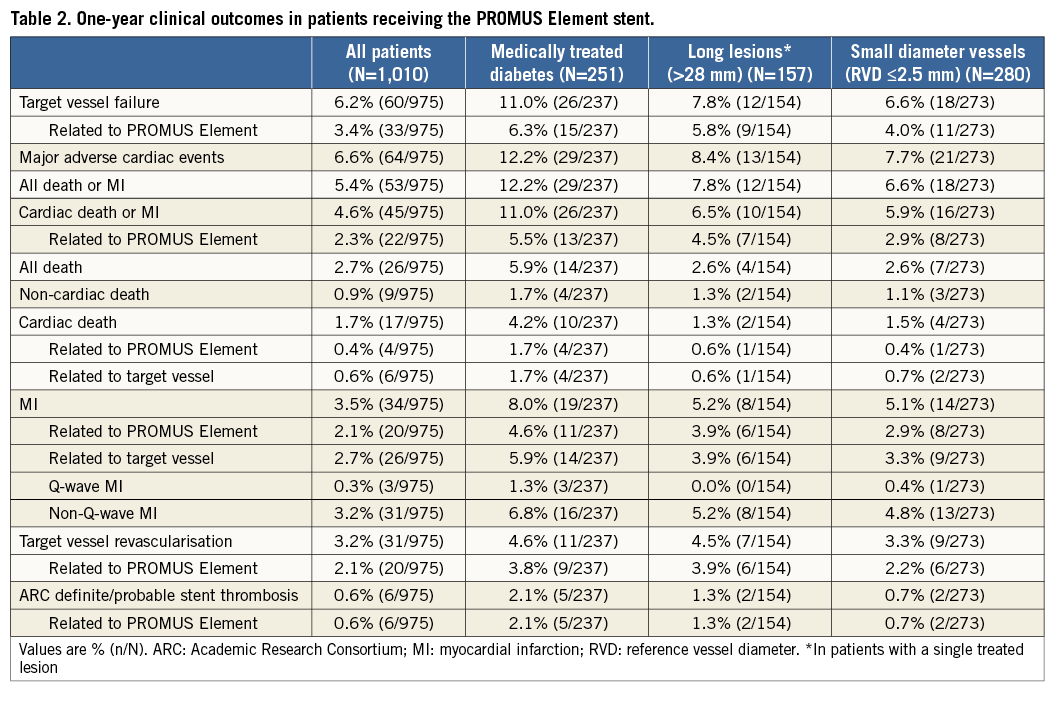
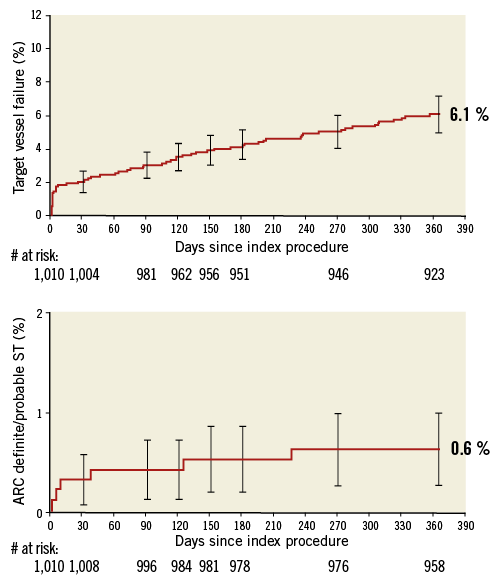
Figure 2. Kaplan-Meier time-to-event analyses baseline to one year. A) Target vessel failure. B) ARC-defined definite/probable stent thrombosis.
At the time of the one-year follow-up visit, 79.6% (756/950) of patients were taking dual antiplatelet therapy, including 96.8% (920/950) on aspirin, 76.1% (723/950) on clopidogrel, 6.2% (59/950) on prasugrel, and 0.1% (1/950) on ticagrelor.
PREDICTORS OF TARGET VESSEL FAILURE AT ONE YEAR
Multivariate predictors of TVF within one year of stent implantation are shown in Online Figure 1. Diabetes, post-procedure dilation, and prior PCI were identified as predictors of both overall TVF and PROMUS Element stent-related TVF. The strongest predictor of overall TVF was the use of thienopyridines for six months or less, and the strongest predictor of TVF associated with the PROMUS Element stent was implantation in a vein graft.
Discussion
In this large, “real-world” population, one-year rates of cardiac death, MI, TVR, and stent thrombosis related to the PROMUS Element stent were low and consistent with rates in the randomised controlled PLATINUM trial of this stent1. Overall, clinical event rates for patients with small calibre vessels (≤2.5 mm) were similar to the rates in the study population as a whole.
As might be expected, medically treated diabetes and single lesion length >28 mm were each associated with modestly increased rates of cardiac events. The technical success rate of 99.8% reflects good deliverability of this stent, which is perhaps a reflection of the conformability and flexibility of this stent platform.
DES IN ROUTINE PRACTICE
This study contributes to a vast and growing body of “real-world” data on contemporary DES outcomes. Studies of DES that have used broad inclusion criteria or an all-comers approach include the PEXIP study of the XIENCE Prime and PROMUS Element everolimus-eluting stents3, the Swedish Coronary Angiography and Angioplasty Registry (SCAAR) analysis of the PROMUS Element stent relative to other contemporary DES4, the XIENCE V USA study of the XIENCE V stent5, the PROENCY registry of the PROMUS everolimus-eluting stent, CYPHER sirolimus-eluting stent, and ENDEAVOR zotarolimus-eluting stent6, the COMPARE study of the PROMUS everolimus-eluting stent and the TAXUS Liberté stent7, and three studies of the Resolute zotarolimus-eluting stent, including the RESOLUTE International Registry8, the Resolute All Comers trial9, and the TWENTE trial10, the latter two both evaluating the Resolute stent versus the XIENCE V stent.
One-year clinical event rates with the PROMUS Element stent in this study are well within the one-year ranges reported from these other all-comers DES studies. The PROMUS Element all-cause mortality rate was 2.7% (other studies 1.6% to 2.8%), the rate of MI related to the PROMUS Element was 2.1% (other studies 0.7% to 7.9%), and the rate of TVR related to the PROMUS Element was 2.1% (other studies 1.3% to 8.2%)3-10. The 0.6% rate of ARC-defined definite/probable stent thrombosis is in the lower range among the all-comers DES studies (other studies 0.0% to 1.6%)3-6,8-11.
STENT DEFORMATION
The occurrence of two cases of longitudinal stent deformation (change in axial length) out of 1,679 stents placed in this study highlights the low frequency of this procedural complication. These two cases of stent deformation, which were detected by angiography during the index procedure, represent classic examples of stent deformation resulting from interaction between an ancillary device and a stent that were easily treated without severe adverse consequences.
Limitations
As a single-arm registry, this study has inherent limitations. Without a control group, the performance of the PROMUS Element stent cannot be directly compared with other contemporary DES. In addition, the broad inclusion criteria and observational design present challenges for evaluating outcomes across studies in which the “all-comers” design results in diverse patient and lesion characteristics. Finally, clinical follow-up at one year was not available for 3.5% of these unselected patients, mostly due to missed visits. Although this represents an acceptable rate of follow-up for a post-market, all-comers registry, given the very low rate of events at one year with this contemporary stent, the potential effect of these missing data should be taken into account when considering the one-year clinical event rates.
Conclusions
In this large and relatively complex group of “real-world” patients, coronary artery revascularisation with the PROMUS Element everolimus-eluting stent resulted in low clinical event rates consistent with those reported for other contemporary DES.
| Impact on daily practice Although randomised controlled trials remain the gold standard for proving the safety and efficacy of new devices in clinical practice, the strict inclusion and exclusion criteria typically used in these trials mean that the results may not be generalisable to an unselected, “real-world” clinical population. This large, multicentre registry presents valuable information on outcomes from everyday clinical practice with the use of the PROMUS Element stent in more than 1,000 patients in Europe. The consistency of the results from this more complex clinical population with those of the randomised controlled trial provide reassurance to the treating physician of good clinical outcomes with the PROMUS Element stent, regardless of patient comorbidities and anatomic complexity. |
Acknowledgements
The authors would like to thank all of the sites that enrolled patients in the PE-Prove study, as well as the clinical events committee members and the associated research organisations (Online Table 2, Online Table 3). In addition, the authors gratefully acknowledge Laurie LaRusso (Chestnut Medical Communications) and Kristin L. Hood (Boston Scientific Corporation) for assistance with manuscript preparation, Shannon Song (Boston Scientific) and Srikanth Garre (Quintiles) for statistical assistance, and Thomas Naeschen (Boston Scientific) for project management.
Funding
This study was sponsored and funded by Boston Scientific Corporation, Marlborough, MA, USA.
Conflict of interest statement
R. Moreno reports lecture and consulting fees from Boston Scientific, Abbott, Cordis, Medtronic, Terumo, and Biotronik. G. Richardt reports serving on an advisory board and receiving speakers’ honoraria from Boston Scientific. M. Thomas reports serving on an advisory board for Boston Scientific. P. Underwood and K. Dawkins are full-time employees with equity interest in Boston Scientific. The other authors have no conflicts of interest to declare.
Online Appendix
METHODS
PATIENT SELECTION, PROCEDURE, AND FOLLOW-UP
The following procedural data were collected: lesion characteristics, total procedure time, antithrombotic and antiplatelet medications administered, and serious adverse events (SAEs), including serious bleeding according to GUSTO classification12. Patients with unsuccessful implant of a study device were followed only up to hospital discharge following the initial attempted index procedure. Enrolment at 14 centres was eventually capped at approximately 40 patients to prevent skewing the results with overrepresentation from high-volume centres.
STUDY ENDPOINTS
Secondary endpoints included stent thrombosis using Academic Research Consortium (ARC) definitions (i.e., definite or probable), major adverse cardiac events (i.e., cardiac death, MI, TVR), cardiac death, MI, TVR, all deaths, and non-cardiac deaths. Technical success was defined as successful deployment of the PROMUS Element stent to the target lesion without device malfunction.
STATISTICAL METHODS
Patient demographics, clinical history, risk factors, pre- and post-procedure lesion characteristics, procedure characteristics, and outcome variables were summarised using descriptive statistics for continuous variables (mean, standard deviation, number of observations, minimum, and maximum) and frequency tables for discrete variables. Kaplan-Meier plots of time-to-event variables were constructed with 95% confidence intervals. Cox models were performed to identify risk predictors with respect to TVF rates. Backward selection was used to identify significant predictors with the threshold to stay in the model set at 0.10.
RESULTS
LONGITUDINAL STENT DEFORMATION
One case was attributed to a deep-seated guide catheter interacting with the proximal end of a deployed PROMUS Element stent, causing slight longitudinal compression, which was resolved with post-dilation and additional stent deployment, without associated patient injury. In the second case, an LAD/diagonal branch bifurcation intervention, the guidewire was jailed by the proximal end of the stent, leading to subsequent unravelling of the guidewire into a filament proximal to the diagonal. During manoeuvres for successful retrieval of the wire with a snare, the proximal end of the implanted stent was minimally damaged. Post-dilation balloon inflations were performed along the entire stented segment and a stent was placed in the proximal LAD/left main. A small non-Q-wave MI (peak CK-MB 43 U/L, troponin 2.4 ng/mL) occurred post procedure and the patient was discharged two days later. Of these two patients with longitudinal stent deformation, only one experienced a small periprocedural enzyme leak. Neither patient has had any other clinical events reported up to the time this manuscript was published.
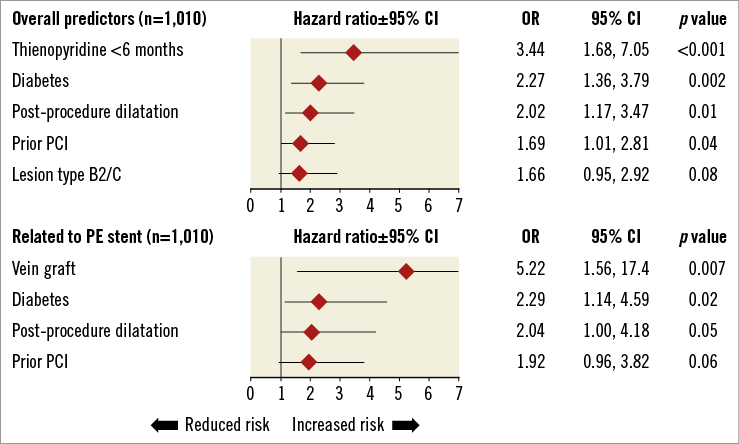
Online Figure 1. Multivariate predictors of target vessel failure within 1 year of stent implantation.