
Abstract
Aims: The EVOLVE China randomised study sought to evaluate the clinical safety and effectiveness of the SYNERGY bioabsorbable polymer-coated everolimus-eluting stent (EES) for the treatment of patients with coronary heart disease in China.
Methods and results: Eligible patients with de novo native coronary artery lesions were randomised (1:1) to receive the SYNERGY or PROMUS Element Plus stent. The primary endpoint was in-stent late loss at nine months. Secondary endpoints included death, MI, revascularisation, and stent thrombosis up to 12 months. A total of 412 subjects were randomised (205 SYNERGY; 207 PROMUS Element Plus) at 14 sites in China from October 2013 to July 2014. SYNERGY was non-inferior to PROMUS Element Plus for the primary endpoint of nine-month in-stent late loss: SYNERGY 0.20±0.33 mm vs. PROMUS Element Plus 0.17±0.38 mm with an upper one-sided 97.5% confidence interval of the difference (0.10 mm), significantly less than the non-inferiority margin (0.15 mm; p<0.0008). Clinical adverse event rates were low and not significantly different between groups at nine and 12 months (all p>0.05).
Conclusions: In the EVOLVE China trial, the SYNERGY bioabsorbable polymer-coated EES was non-inferior to the PROMUS Element Plus permanent polymer-coated EES for the primary endpoint of late loss at nine months.
Abbreviations
BP-BES: bioabsorbable polymer-coated biolimus-eluting stent
DAPT: dual antiplatelet therapy
DES: drug-eluting stent
EES: everolimus-eluting stent
MI: myocardial infarction
QCA: quantitative coronary angiography
ST: stent thrombosis
TLF: target lesion failure
TLR: target lesion revascularisation
TVR: target vessel revascularisation
Introduction
Drug-eluting stents (DES) with permanent polymers reduce the risk of clinical restenosis and the need for repeat revascularisation compared with bare metal stents. However, the permanent polymers, especially those used on first-generation DES, have been associated with chronic inflammation and impaired healing1-3. Biodegradable polymer-coated stents may overcome this problem as the polymer is fully degraded after a few months. The safety and performance of SYNERGY™ (Boston Scientific, Marlborough, MA, USA), a biodegradable polymer everolimus-eluting stent (BP-EES), was evaluated in the first-human-use EVOLVE study and the pivotal, “more-comers” EVOLVE II study4-6. The EVOLVE study found that SYNERGY was non-inferior to PROMUS Element (PE) for six-month in-stent late loss5. The larger EVOLVE II randomised clinical trial demonstrated non-inferiority of SYNERGY to PE Plus for one-year TLF and similar rates of MI and target lesion revascularisation (TLR)4. However, clinical outcomes for the SYNERGY stent have not been evaluated in patients from China. The objective of the EVOLVE China study was to evaluate the safety and effectiveness of the thin-strut, everolimus-eluting, platinum-chromium bioabsorbable polymer-coated SYNERGY stent for the treatment of patients with coronary heart disease in China.
Methods
STUDY DESIGN
The EVOLVE China clinical trial is a prospective, multicentre, single-blind, 1:1 randomised (SYNERGY to PE Plus), controlled, non-inferiority trial.
DEVICE DESCRIPTION
The investigational device in EVOLVE China was the SYNERGY stent, a thin-strut platinum-chromium stent coated on the abluminal side with an ultrathin 4 mm bioabsorbable polymer, poly(D,L-lactide-co-glycolide) (PLGA), which elutes everolimus4,5. The PE Plus is a thin-strut platinum-chromium stent coated with a permanent polymer eluting everolimus7,8.
SUBJECT SELECTION, PROCEDURE, AND FOLLOW-UP
The EVOLVE China study is registered at www.clinicaltrials.gov, identifier NCT01966159. Clinical criteria included age 18 to 75 years with symptomatic one- or two-vessel coronary artery disease with objective evidence of ischaemia or silent ischaemia, and a left ventricular ejection fraction >30%. Lesion criteria included de novo lesions in native coronary arteries with diameter ≥2.25 mm to ≤4.0 mm, ≤34 mm long with stenosis ≥50% to <100% and TIMI flow >1 (and one of the following: ≥70% stenosis, abnormal fractional flow reserve, abnormal stress or imaging stress test, or mildly elevated biomarkers prior to the procedure). Patients with acute MI (with documented elevation in cardiac enzymes), left main disease, chronic total occlusions, vein graft disease, in-stent restenosis, complex bifurcations (those requiring treatment with >1 stent), or target vessels treated with any type of percutaneous coronary intervention (PCI) in the past 12 months were excluded per protocol.
Balloon catheter predilation was required. Angiography was performed using standard techniques; two orthogonal projections of the normal reference segment and stenosis in an unforeshortened view were required. Intracoronary nitroglycerine was given prior to imaging. Protocol-mandated angiographic nine-month follow-up was required at the same study centre that performed the baseline assessment. Stent implantation was performed according to standard of care at each institution. Dual antiplatelet therapy (DAPT) with aspirin and a P2Y12 inhibitor was prescribed post PCI for six months (12 months in patients at low risk of bleeding). All patients provided written informed consent before enrolment.
DATA MANAGEMENT
Study monitors verified all case report form data. Clinical events were adjudicated by an independent clinical events committee. An independent data safety monitoring committee evaluated all reported and adjudicated adverse events at regular intervals. Angiographic data were analysed by an independent core angiographic laboratory (CCRF Co. Ltd, Beijing, China).
Clinical follow-up occurred in-hospital, at 30 days, 6 months, 9 months, and 12 months, and will occur annually for five years. Enrolled subjects who did not receive a study stent were followed up to 12 months.
ENDPOINTS
The primary endpoint was in-stent late loss at nine months post index procedure (by quantitative coronary angiography [QCA]). Secondary endpoints included death, MI, revascularisation, and ST. Spontaneous MI was defined as the rise and/or fall of cardiac biomarkers with ≥1 value >99th percentile of the upper reference limit (URL) and with evidence of myocardial ischaemia.
Periprocedural MI was diagnosed if at least one of the following occurred: 1) CK-MB >3x the URL without clinical or imaging correlates, 2) new pathologic Q-waves, or 3) autopsy evidence of acute MI4. Post-procedure enzyme collection was mandated. ST was defined according to the Academic Research Consortium (ARC) definitions9.
The secondary angiographic endpoints included % diameter stenosis (DS), binary restenosis, and minimum lumen diameter (MLD). Technical success was defined as successful delivery and deployment of the study stent to the target vessel, without balloon rupture, or stent embolisation. Clinical procedural success was defined as mean lesion diameter stenosis <30% in two near orthogonal projections with TIMI 3 flow (visual assessment) without in-hospital MI, TVR, or cardiac death.
STATISTICAL METHODS
Endpoints were analysed on an intention-to-treat (ITT) and on a per-protocol basis; the per-protocol population was the primary analysis set for the non-inferiority assessment. The sample size needed to evaluate non-inferiority was based on an expected mean difference in nine-month in-stent late loss between groups of 0 mm, an expected common standard deviation of 0.4 mm, significance level of 2.5% (one-sided), and a non-inferiority margin of 0.15 mm. An estimated 200 patients in each arm (after accounting for 10% attrition) resulted in >90% power. Non-inferiority was concluded if the one-sided upper 97.5% confidence bound for the difference in nine-month in-stent late loss between groups was less than the non-inferiority margin corresponding to p<0.025 from a one-sided Student’s t-test. Clinical event rates were summarised by treatment group using proportions and continuous data means, standard deviations, and sample sizes. Binary rates were compared with a chi-square or a Fisher’s exact test. A Student’s t-test was used to assess differences between treatment groups for continuous endpoints. Kaplan-Meier time-to-event plots were constructed for clinical events; treatment groups were compared using the log-rank and Wilcoxon tests. Univariate and multivariate analyses were performed to assess the effect of possible predictors on the primary endpoint of nine-month in-stent late loss. For multivariate analysis, baseline and procedural variables were included in a univariate logistic regression model and then, in a stepwise fashion, included in the multivariate regression model.
The significance level thresholds for entry and exit of independent variables were set at 0.1. Statistical analysis was performed using SAS software, version 9.1.3 (SAS Institute, Cary, NC, USA).
Results
PATIENT DISPOSITION, BASELINE, AND PROCEDURAL CHARACTERISTICS
In total, 412 subjects were randomised at 14 sites in China between October 2013 and July 2014 (PE Plus n=207 and SYNERGY n=205). At nine months, angiographic data were available in 90.7% and 87.0% of SYENRGY and PE Plus patients, respectively (Figure 1). At 12 months, clinical follow-up was available in 97.1% of SYNERGY and 99.0% of PE Plus patients (Figure 1).
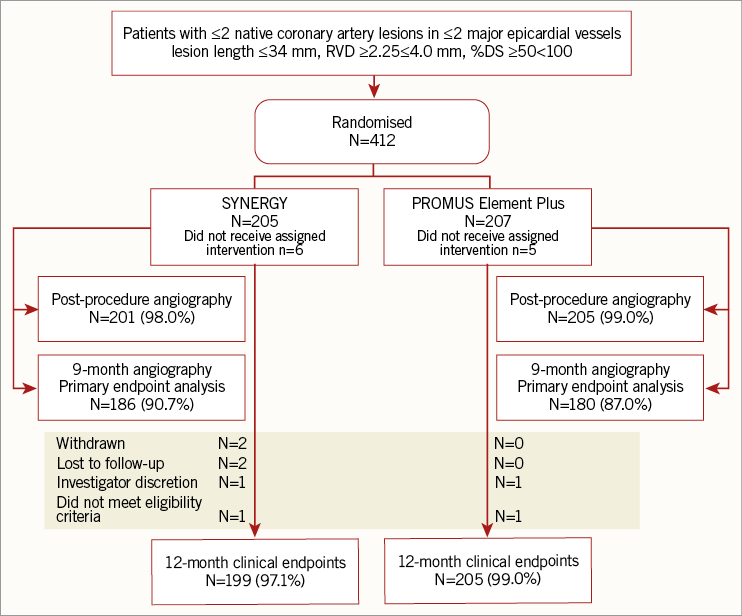
Figure 1. Patient disposition in EVOLVE China.
Baseline clinical characteristics were well balanced between treatment arms. The average age of patients was 58 years, 70% were male, and 30% had diabetes (Table 1). Baseline lesion characteristics were also similar between groups, except a slightly larger baseline MLD in SYNERGY compared to PE (Table 3).
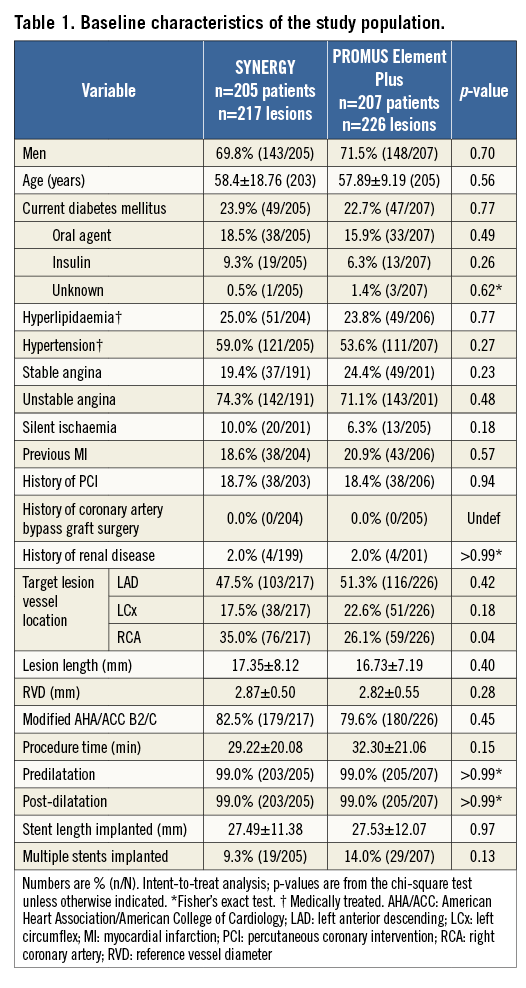
The rates of technical and clinical procedural success were not significantly different between treatment arms (technical success: SYNERGY 99.5%, PE Plus 99.2%, p>0.99; clinical procedural success 98.0% vs. 98.0%, p>0.99, respectively).
Aspirin usage was high and not significantly different between groups up to 12 months (discharge: SYNERGY 99.5%, PE Plus 100%, p=0.50; 12 months: SYNERGY 96.5%, PE Plus 97.5%, p=0.53). DAPT usage at discharge and 12 months was also high (discharge: SYNERGY 97.5%, PE Plus 98.5%, p=0.50; 12 months: SYNERGY 86.4%, PE Plus 88.2%, p=0.59).
ANGIOGRAPHIC OUTCOMES AT 9 MONTHS
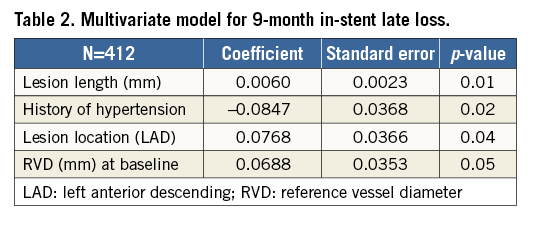
The primary endpoint of nine-month in-stent late loss in SYNERGY was non-inferior to PE Plus (Figure 2A). The absolute difference between treatment groups was 0.03 mm (SYNERGY 0.20±0.33 mm; PE Plus 0.17±0.38 mm) with an upper one-sided 97.5% confidence interval of 0.10 mm, significantly less than the non-inferiority margin of 0.15 mm (p<0.0008). Outcomes analysed in the ITT patient population were very similar. The cumulative distribution function for in-stent late loss at nine months is shown in Figure 2B. Multivariate predictors of nine-month in-stent late loss included longer lesion length, LAD lesion location, and larger reference vessel diameter; a history of hypertension was associated with lower in-stent late loss (Table 2).
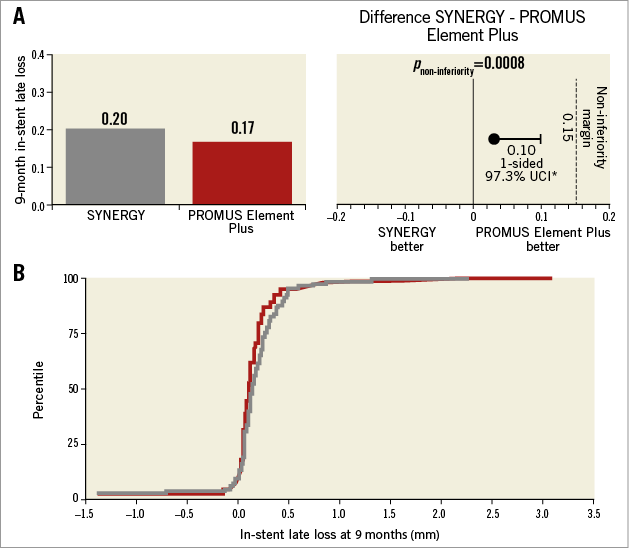
Figure 2. Primary endpoint. The primary endpoint, the difference in nine-month in-stent late loss between treatment groups (per patient), was less than the performance goal (0.15); specifically, the one-sided upper 97.5% confidence bound for the difference was <0.15 mm in both the per-protocol and ITT populations. A) Per-protocol patient population. B) Cumulative in-stent late loss at nine months in SYNERGY (grey) and PROMUS Element Plus (red).
Additional QCA outcomes at nine months are shown in Table 3. SYNERGY was similar to PE Plus with regard to nine-month MLD, binary restenosis and late loss (Table 3). In-stent % DS was higher in the SYNERGY arm at nine months (SYNERGY 12% vs. PE Plus 9%, p=0.02), but was similar when the analysis segment was used (20% vs. 19%, p=0.74; Table 3). The non-paired analysis was nearly identical to the paired analysis.
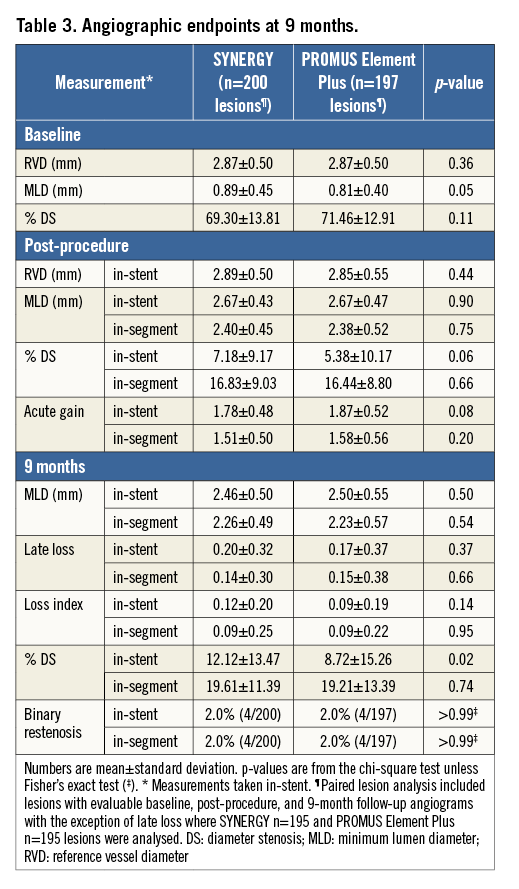
CLINICAL OUTCOMES AT 12 MONTHS
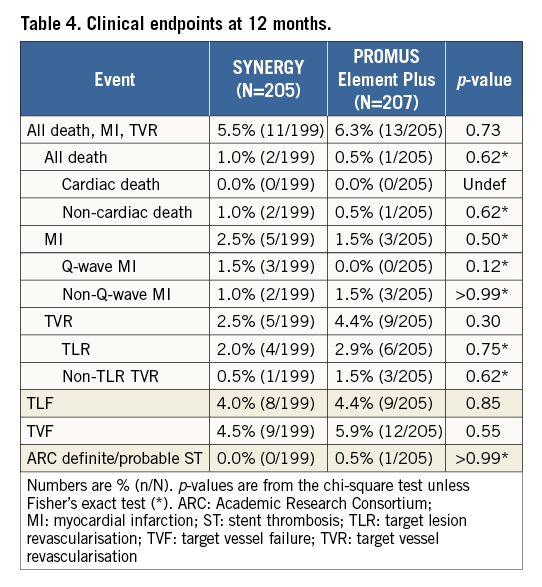
Clinical event rates were low and not significantly different at nine months; these low rates were sustained at 12 months (Table 4). At 12 months, two patients in the SYNERGY arm and one patient in the PE arm had a non-cardiac death. MI occurred in 2.5% of SYNERGY patients and 1.5% of PE Plus patients (p=0.50). TLR and TLF at 12 months were not significantly different between groups (Table 4). No ST occurred in the SYNERGY arm; one PE Plus patient experienced a definite ST on the day of the index procedure.
Discussion
In this randomised EVOLVE China trial, the SYNERGY stent was non-inferior to the PE Plus stent for in-stent late loss at nine months. Furthermore, the rates of TLF were low and similar for both stents; no ST was reported in the SYNERGY arm at one year. Angiographic outcomes with SYNERGY were previously examined in the EVOLVE II QCA study, which found similar in-stent late loss at nine months (0.22±0.33 mm) to the SYNERGY arm of the EVOLVE China (Meredith et al. http://www.crtonline.org/presentation-detail/nine-month-primary-endpoint-results-of-evolve-ii-q). Late loss was comparable to other BP-DES (range 0.10 to 0.27 mm over eight to nine months) as well as other permanent polymer EES (range: 0.10 to 0.30 mm over eight to nine months)10-21. The EVOLVE II trial demonstrated that SYNERGY was non-inferior to the PE Plus stent for TLF at one year; short- and mid-term clinical results were also similar4.
Metal stents have improved in terms of strut thickness, material, and polymer type. Many first-generation polymers have proinflammatory properties leading to incomplete healing and delayed endothelial coverage, potentially contributing to late events including ST and restenosis22,23. A promising strategy to reduce long-term events further is the use of BP-DES which leaves behind a bare but inert and biocompatible metal after polymer degradation. This limits the length and amount of polymer exposure. In animal studies, PLGA absorption and drug release from the SYNERGY stent are complete shortly before four months6. Preclinical work found that BP-DES were associated with a reduction in the inflammatory response to stent implantation and had more rapid neointimal coverage compared to permanent polymer DES in pigs and rabbits24-26. In humans, two small single-arm optical coherence tomography (OCT) studies found almost complete coverage of the stent by three months in vivo27,28.
Most clinical benefits of a polymer which degrades would be anticipated to accrue slowly over time after stent implantation. If late events such as ST were reduced, the length of DAPT duration required after stent implantation could be shortened. Objectively, the value of BP-DES has not yet been proven by a significant reduction in long-term clinical events. Outcomes after implantation of current-generation permanent polymer DES in conjunction with proper adjunctive medications are good with very low rates of death, MI and ST up to five years29. BP-DES have shown similar safety and effectiveness compared to second-generation DES long-term; low rates of ST after implantation have been found with SYNERGY and other new-generation BP-DES10,30. At three years, MACE and its components as well as ST were not different between BP biolimus-eluting stents (BES) and permanent polymer EES in the COMPARE II trial31. Five-year outcomes between BP-BES and permanent polymer EES were similar in the ISAR-TEST-4 trial32. Since few long-term studies of thin-strut BP-DES have been published, differences between these BP-DES and permanent polymer DES may be detected at later follow-up times. Additionally, expected outcomes after treatment with biodegradable polymers may not be a class effect. There are important differences in strut thickness, metal and geometry, antiproliferative drug, polymer degradation, and the drug elution profile between BP-DES. A head-to-head trial comparing two BP-DES, a thin-strut cobalt-chromium sirolimus-eluting stent versus a stainless steel thicker strut biolimus-eluting stent, found significant differences in the risk of ST at one year33.
In a recent meta-analysis, BP-DES treated patients had significantly lower late lumen loss and late ST compared to permanent polymer DES treated patients11. However, when grouped into first- and second-generation DES, late ST after BP-DES occurred less often compared to first-generation DES but at a similar rate compared to second-generation DES. A large network meta-analysis found higher rates of definite ST with BP-BES compared to newer-generation cobalt-chromium permanent polymer EES29. Newer BP-DES, including SYNERGY, were not included in these meta-analyses. A recent meta-analysis demonstrated equivalent or lower rates of definite/probable ST at one year with SYNERGY compared to other stents and bioabsorbable scaffolds34.
Limitations
This study has several limitations: (1) by reason of its single-blind design, the implanting physicians were aware of the stent being deployed; (2) the study was not powered for clinical endpoints; however, the EVOLVE II trial was powered for 12-month TLF that showed non-inferiority of SYNERGY versus the PE Plus; (3) similar to most randomised controlled trials, the use of inclusion and exclusion criteria may limit the generalisability of these results to patients in real-world clinical practice.
Conclusions
The SYNERGY bioabsorbable polymer-coated EES was non-inferior to the PROMUS Element Plus permanent polymer-coated EES for nine-month in-stent late loss in EVOLVE China. Low adverse clinical event rates up to one year with no ST were observed in the SYNERGY arm. These data support the safety and efficacy of the SYNERGY stent for the treatment of Chinese patients with stable or unstable coronary artery disease.
| Impact on daily practice Although improvements in stent material, geometry, thickness, and polymer biocompatibility have reduced adverse clinical event rates compared with early-generation DES, there remain concerns regarding neoatherosclerosis and late events. Recently, the thin-strut, everolimus-eluting, platinum-chromium bioabsorbable polymer-coated SYNERGY stent demonstrated comparable midterm rates of TLF when compared with a permanent polymer everolimus-eluting stent. The objective of the EVOLVE China study was to evaluate and confirm the safety and effectiveness of SYNERGY for the treatment of patients with coronary heart disease in China. Longer-term follow-up of SYNERGY in real-world patient populations is needed. |
Acknowledgements
We thank Kristine Roy, PhD, for assistance in manuscript preparation and Peter Lam for statistical analysis (both Boston Scientific Corporation).
Funding
This study was sponsored by Boston Scientific.
Conflict of interest statement
Y. Han has received research grants from Boston Scientific, Essen (Beijing, China), JWMS (Shandong, China) and Lepu Medical (Beijing, China). D. Allocco, M. Zhang and K. Dawkins are full-time employees and stockholders of Boston Scientific. The other authors have no conflicts of interest to declare.
