Abstract
Transcatheter aortic valve implantation (TAVI) is a relatively new technique that has been introduced to treat inoperable and high-risk patients with severe aortic stenosis. From its early stages it became apparent that TAVI has tremendous potentialities and thus a considerable effort was made to design new prostheses and advance TAVI technology that would make easier and feasible its application in complex anatomies and in patients with multiple comorbidities. In addition, evidence from randomised control trials have emerged demonstrating that it improves prognosis in inoperable patients (PARTNER trial cohort B) and that it can be considered as an attractive alternative to surgery in patients with a high operative risk (PARTNER trial cohort A). These encouraging data have motivated the scientific community to organise further trials, which will examine the performance of new devices and explore the feasibility of TAVI in different groups. In this article we review the literature, present the advances in TAVI technology, cite the evidence from the already published studies and discuss the upcoming clinical trials.
Introduction
Transcatheter aortic valve implantation (TAVI) is a relatively new technique introduced in 2002 by Cribier to treat patients who suffer from severe aortic stenosis1. The encouraging results from its first applications as well as the prospect of a less invasive valve replacement strategy has drawn the attention of the scientific community. Hence, over the last few years a whole industry has been developed around TAVI and a considerable effort has been made to create new valves and enabling TAVI devices that will overcome the limitations of the first-generation devices, reduce the risk of complications and increase TAVI applications. In addition, several trials have been organised to examine the performance of these innovations and explore the potentialities of TAVI in different populations. In this review article we present the new TAVI devices, cite the current evidence and discuss the ongoing/upcoming clinical trials.
Transcatheter devices
COMMERCIALLY-AVAILABLE TECHNOLOGY
EDWARDS SAPIEN™
There are two valves in widespread use: the balloon-expanding Edwards SAPIEN transcatheter valve (THV) (Edwards Lifesciences, Irvine, CA, USA) and the self-expanding CoreValve® (Medtronic Inc., Minneapolis, MN, USA). The Edwards SAPIEN THV has been designed for transapical and transfemoral implantation and incorporates bovine pericardial leaflets sutured onto a stainless steel stent (Figure 1). Transfemoral implantation is performed using the RetroFlex™ delivery system (Edwards Lifesciences, Irvine, CA, USA), which has a large diameter (22-24 Fr depending on the size of the valve), while the transapical implantation utilises a 26 Fr delivery sheath. The success rate with this device is high (95.2% for the transfemoral and 92.7% for the transapical approach) as it has been reported in the SOURCE registry that included 1,038 patients2. The mortality rate at 30 days was higher in the patients who underwent transapical valve implantation (10.3% vs. 6.3%) and these patients also had a worse one-year prognosis (72.1% vs. 81.1%) probably because of the increased comorbidities3. There was no difference between the two groups in the rate of stroke and the need for permanent pacemaker implantation but on the other hand a five-fold higher vascular complication rate was noted in patients who had transfemoral valve implantation (22.9% vs. 4.7%), a fact that was attributed to the large diameter of the RetroFlex sheath.
This limitation was addressed by the Edwards SAPIEN XT™ (Edwards Lifesciences, Irvine, CA, USA) device, which is an updated version of the SAPIEN THV. The Edwards SAPIEN XT has a cobalt chromium frame with struts that are thinner and have a more open structure. These modifications provide the necessary stability and radial force and allow a tighter crimping. Similarly to Edwards SAPIEN THV, the leaflets of the valve are made from bovine pericardium, but they have a different scallop design and arrangement. In particular they are semi-closed when the valve is in its natural position, a fact which reduces the time and the pressure difference required to close the valve during early diastole (Figure 1). Finally, in contrast to the Edwards SAPIEN THV, which is crimped directly over the balloon, the SAPIEN XT has a specially designed delivery system (NovaFlex™; Edwards Lifesciences, Irvine, CA, USA), which allows the valve to be mounted onto the balloon when it is in the abdominal aorta. The NovaFlex system consists of two catheters (outer and inner balloon catheters). Initially the valve is positioned on the shaft of the outer catheter proximally to the balloon and when the device is in the abdominal aorta the balloon catheter is retracted, while the outer catheter remains fixed. Thus, the balloon is pulled towards the valve4,5. Optimal alignment is facilitated via two radiopaque markers located at the proximal and distal end of the balloon. These modifications allow valve implantation through a lower profile delivery system (external diameter, 18-19 Fr, depending on the size of the valve) and have reduced the risk of vascular complications. Indeed in a recent study performed in 120 patients there was a three-fold lower vascular event rate (11.1% vs. 33%; p=0.004) in the group of patients treated with the Edwards SAPIEN XT compared to those who received an Edwards THV valve6. The Edwards SAPIEN XT is nowadays preferred over the SAPIEN THV and currently two large studies are being conducted with this device: the Placement of Aortic Transcatheter Valves II (PARTNER II) trial, which will be described in the following section, and the Aspirin Versus Aspirin and ClopidogRel Following Transcatheter Aortic Valve Implantation (ARTE) trial, which aims to compare the efficacy of aspirin vs. the combination of aspirin and clopidogrel in preventing major ischaemic events (Table 1).
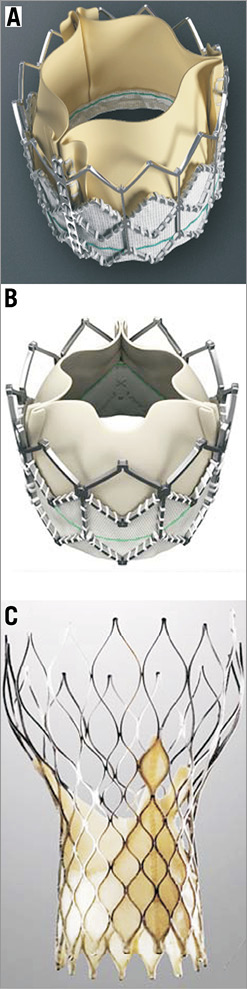
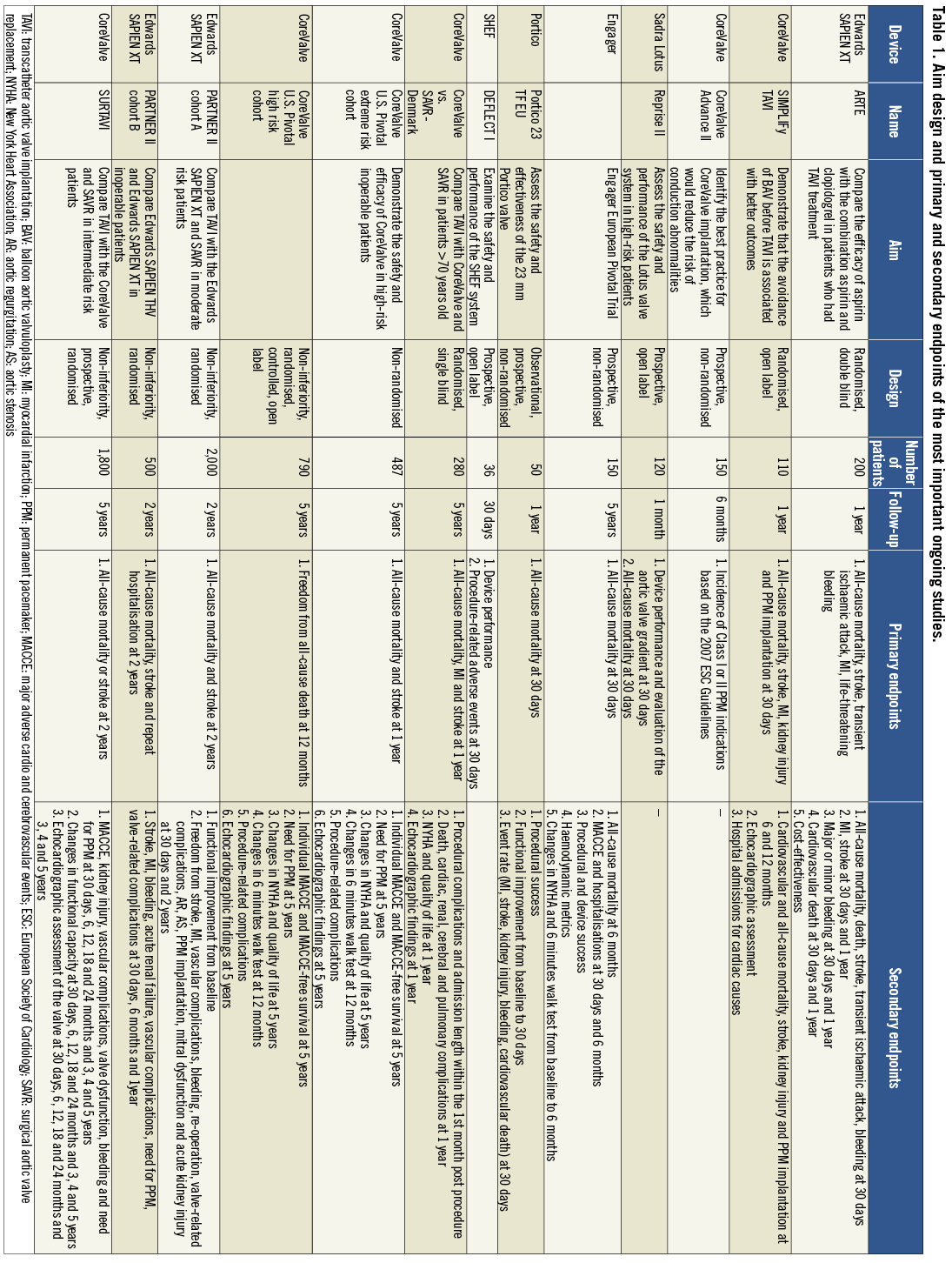
Figure 1. First-generation devices: A) Edwards SAPIEN THV (Edwards Lifesciences, Irvine, CA, USA); B) Edwards SAPIEN XT (Edwards Lifesciences, Irvine, CA, USA); C) CoreValve (Medtronic Inc, Minneapolis, MN, USA). The image was modified and reproduced with permission31.
COREVALVE®
The other commercially available valve is the CoreValve® (Medtronic Inc., Minneapolis, MN, USA), which consists of porcine pericardial leaflets mounted onto a self-expanding nitinol frame (Figure 1). The design of the valve does not allow antegrade implantation but on the other hand it has the advantage that it uses a lower profile delivery system AccuTrack™ (Medtronic Inc.) 18 Fr compared to the Edwards SAPIEN THV prosthesis. Thus, in the Medtronic CoreValve multicentre expanded evaluation registry the vascular access complication rate was only 1.9% while in the ADVANCE registry, which included data from 1,015 patients, it was 10.7%7,8. The reported mortality rate at 30 days in the ADVANCE registry was 4.5%; 2.9% of the patients had a stroke and 26.3% required a pacemaker implantation. The low profile of the CoreValve seems also to facilitate device implantation without balloon predilation as has recently been demonstrated in a small feasibility study9. It has been speculated that direct valve deployment may contribute to a smaller risk of complications as most of them (e.g., stroke, conduction abnormalities and paravalvular leaks) are caused during the manipulations of the device in the aortic valve, its annulus and the left ventricular outflow tract. Thus, the SIMPLIFy TAVI study has been designed to investigate whether the avoidance of balloon valvuloplasty reduces morbidity during TAVI. The study aims to randomise 110 patients with a logistic EuroSCORE of ≥15% and poor left ventricular ejection fraction (≤35%) to TAVI with and without balloon valvuloplasty. Randomisation has already started and the trial is expected to be reported within this year.
The higher risk for permanent pacemaker implantation that has been noticed after CoreValve implantation has been attributed to the fact that the valve is situated lower in the left ventricular outflow track than the Edwards SAPIEN THV, and thus it is possible to traumatise the atrioventricular node and the bundle of His10. Several studies have tried to identify predictors of new conduction abnormalities and currently the CoreValve Advance II prospective registry is underway, which aims to define the best practice for CoreValve implantation that may reduce the need for permanent pacemaker implantation11,12.
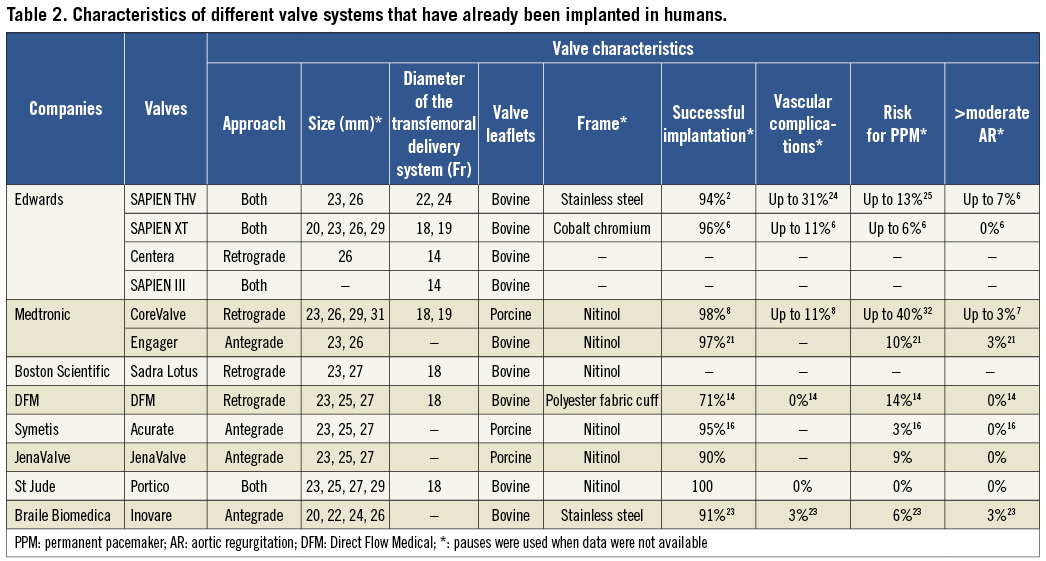
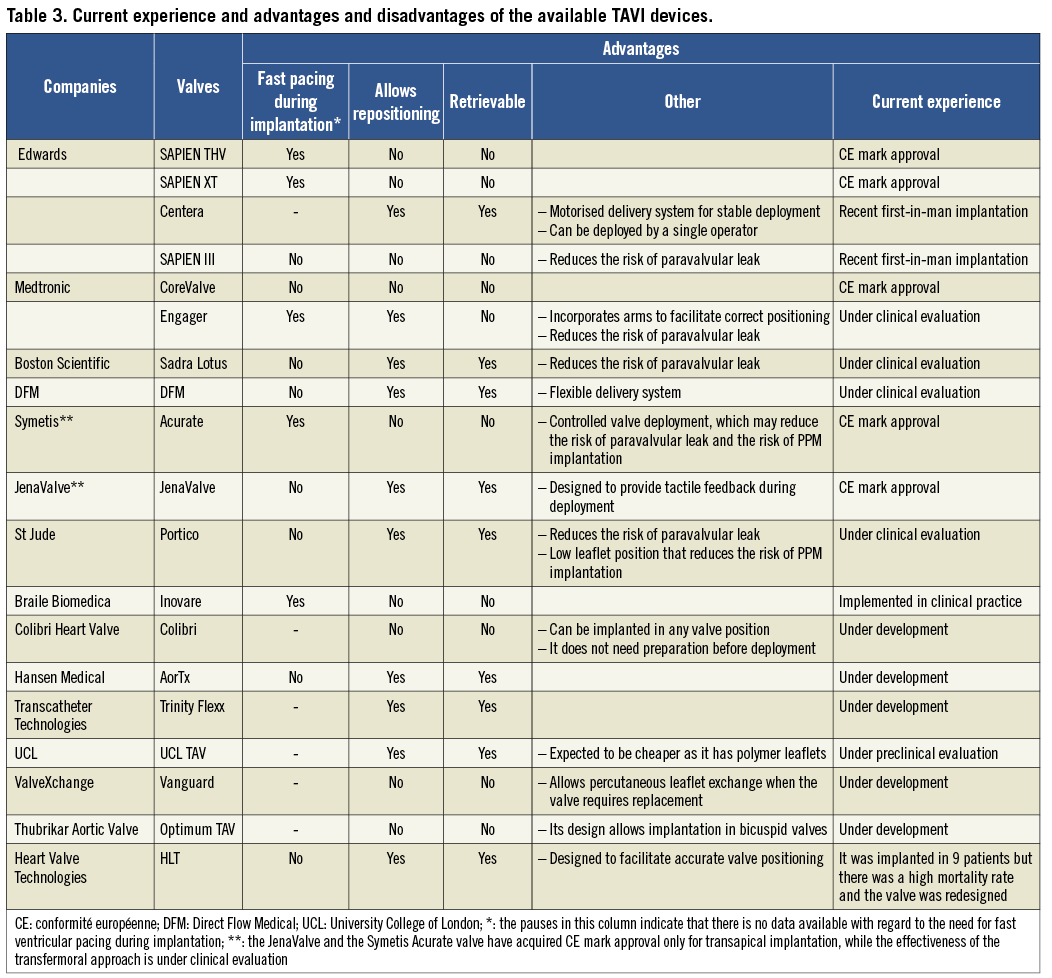
Finally, a significant limitation that all the first-generation valves have is the fact that they cannot be retrieved or repositioned after implantation and thus in case of erroneous placement a second transcatheter valve has to be deployed or conventional surgery with sternotomy and valve replacement is needed as a bail-out procedure. To address all the above-mentioned drawbacks, several new valve systems have been developed (Figure 2, Table 2). Some of them have already proven their effectiveness while others are currently under evaluation (Table 3).

Figure 2. Second-generation TAVI devices: A) DFM (Direct Flow Medical Inc, Santa Rosa, CA, USA); B) HLT valve (Heart Valve Technologies Inc, Maple Grove, MN, USA); C) Inovare (Braile Biomedica, Sao Paulo, Brazil); D) JenaValve (JenaValve, Munich, Germany); E) Portico valve (St Jude Medical, St Paul, MN, USA); F) Sadra Lotus (Boston Scientific, Natick, MA, USA); G) Acurate valve (Symetis SA, Ecublens, Switzerland); H) Engager (Medtronic Inc, Minneapolis, MN, USA); I) UCL TAV valve (University College of London, London, UK); J) Vanguard II valve (ValveXchange Inc, Greenwood Village, CO, USA); K) Trinity TriFlexx (Transcatheter Technologies, Regensburg, Germany); L) the AorTx valve (Hansen Medical Inc, Mountain View, CA, USA). The figure is a combination of the images obtained from Rodes-Cabau and Chiam and Ruiz and have been reproduced with permision32,33.
SECOND-GENERATION VALVES
SADRA LOTUS VALVE
The Sadra® Lotus™ valve (Boston Scientific, Natick, MA, USA) is the first second-generation valve and was implanted for the first time in man in 200813. It consists of three bovine pericardial leaflets that are attached to a self-expanding nitinol frame that is surrounded in its lower part by a sealing membrane that reduces the risk of paravalvular leak. The valve had initially seven, but in the second generation it has three, locking mechanisms that allow control of the implantation and incorporates fluoroscopic markers to guide correct placement. Another advantage of the device is that it permits repositioning and it is fully retrievable. It can be placed only through the retrograde approach with the use of a 21 Fr delivery system for the initial Sadra Lotus, and an 18 Fr delivery system for the redesigned valve.
The performance and effectiveness of the valve is currently under investigation in the Reprise I feasibility study, which is a small non-randomised open label trial that aims to recruit 10 patients and is going to be reported at the cardiovascular course, EuroPCR, in 2012. The Reprise II study is expected to start a few months later and intends to include 120 patients, in 15 European centres, and will be conducted with the aim to provide a conformité européenne (CE) mark approval for the device.
DIRECT FLOW MEDICAL VALVE
The Direct Flow Medical (DFM) valve (Direct Flow Medical Inc, Santa Rosa, CA, USA) is the first non-metallic valve, and was introduced four years ago. It incorporates three bovine pericardial leaflets that are encased in an inflatable framework with a slightly tapered polyester fabric cuff. In the upper and lower margin of the cuff there are two inflatable rings that are connected to three detachable positioning and fill lumens that are used to inflate and deflate the valve. The valve can only be deployed transfemorally through a 22 Fr delivery system, which has proximal end handles to control valve positioning. Its design allows repositioning and even full retrieval before being deployed. A small feasibility study conducted in 15 patients provided promising results reporting low periprocedural mortality (one event due to myocardial infarction caused by coronary occlusion after valve deployment) and morbidity (one stroke) and highlighted the advantages of this device, which provides the operator with unprecedented freedom in the handling of the valve during implantation14. The recently reported two-year follow-up showed that the non-metallic valve has a stable haemodynamic performance and maintains its position and shape with no evidence of recoil15. The company is planning to organise a larger feasibility study, the “Direct Flow 18 Fr CE Mark trial”, which will investigate the effectiveness of the valve in high-risk patients to enable the valve to acquire CE mark approval.
SYMETIS® ACURATE™ VALVE
Another recently introduced prosthesis is the Symetis® Acurate™ valve (Symetis SA, Ecublens, Switzerland) , which was designed to facilitate optimal positioning. The valve can be implanted through the transapical approach using a sheathless implantation system with a 28 Fr diameter. It consists of porcine tissue leaflets that are attached to a self-expanding nitinol stent. This frame is connected to three arches that provide better stability during implantation, while its distal edge forms a crown that is designed in such a way as to provide axial fixation and tactile feedback during positioning. A recent feasibility study performed in 40 patients demonstrated a success rate of 95%, while the 30-day mortality rate was 12.5% and was due mainly to respiratory dysfunction (three out of the five patients). Two patients experienced a stroke and only one patient required pacemaker implantation16. These results were similar to those reported in a larger group of patients (90 subjects) presented recently in the Transcatheter Cardiovascular Therapeutics meeting TCT (2011) in which the mortality rate was only 7.8% and there was no significant aortic regurgitation17. The company also plans to evaluate the safety and the feasibility of the Acurate™ valve implantation through the transfemoral approach. The study, called “Acurate TF™ first-in-man”, is expected to start this year. In this non-randomised, non-controlled, prospective open label study, 20 patients will be recruited and they will be followed up for five years.
JENAVALVE
JenaValve (JenaValve, Munich, Germany) is another device that has been developed to allow more controlled and anatomically correct positioning. The three leaflets of the valve are made from porcine tissue and are attached to a self-expanding nitinol frame that has three feelers which are designed to embrace the native valve leaflets during implantation18. This mechanism is expected to provide tactile feedback during valve positioning and allow co-axial valve deployment. In addition, the valve permits repositioning and it is fully retrievable. A small feasibility study has recently been conducted which examined the safety and the performance of the valve in 67 patients. In all cases the JenaValve was implanted through the transapical approach although it can be deployed transfermorally as well. The results were reported at the European Association for Cardio-Thoracic Surgery in 2011 and the PCR London Valves 2011 meetings19,20. The mortality rate at 30 days post operation was only 7.6% (3% due to cardiac reasons), the stroke rate was 3%, 9.1% of the patients required a pacemaker implantation and there was no significant aortic regurgitation. Based on these results the valve received CE mark approval for transapical implantation. The company is planning to organise another study, the “JenaValve transfemoral first-in-man”, which will start in mid-2012 and will examine the safety and feasibility of the valve through the transfemoral approach.
ENGAGER™ VALVE
The Engager™ valve (Medtronic Inc., Minneapolis, MN, USA) is a self-expanding valve and consists of three bovine pericardial leaflets sewn to a polymer sleeve. This is mounted onto a nitinol frame with sinus support arms designed to allow anatomically-correct positioning and deployment. The valve is implanted through the transapical approach using a 30 Fr delivery system. It allows re-positioning under fluoroscopic guidance but it is not retrievable. In a recent feasibility study conducted in 30 patients the valve was successfully implanted in 97% of the subjects but there was a high rate of aortic dissections (13%) and an increased 30-day mortality (20%), which was at least partially attributed to the rigid delivery system used for valve deployment21. This elicited a thorough review of the valve and delivery system, which has been redesigned. Currently the Engager™ European Pivotal trial is underway, which aims to examine the safety and the performance of the improved device (Table 1).
PORTICO™ VALVE
The Portico™ valve (St Jude Medical, St Paul, MN, USA) is a new device designed in such a manner as to allow single operator valve deployment. It consists of a nitinol self-expanding stent which has a tissue cuff that minimises the risk of paravalvular leak. The bovine leaflets are placed close to the ventricular end of the stent in order to reduce valve protrusion into the left ventricular outflow tract. The valve has been designed to allow both transapical and transfemoral implantation using a 24 Fr and 18 Fr delivery system, respectively, and does not require fast ventricular pacing during implantation. One significant advantage of the Portico valve is that it can be repositioned and retrieved until fully deployed. The performance of the valve has been recently evaluated in a small feasibility study that included 10 patients –all females– who underwent successful valve implantation through the transfemoral approach22. At 30-day follow-up there were no major adverse events, no significant aortic regurgitation and none of the patients required pacemaker implantation. Currently, the Portico 23TF EU feasibility study is underway, which aims to assess the safety and the effectiveness of the delivery system and the 23 mm Portico valve in 50 patients with severe aortic stenosis. The patients will be followed for one year and the study is expected to be completed by the end of 2012.
OTHER VALVES
The Inovare® valve (Braile Biomédica, Sao Paulo, Brazil) is a balloon-expandable bioprosthesis designed for retrograde implantation. It has similar limitations to the first-generation valves and was developed in Brazil to satisfy the national needs, reduce the cost and broaden the applications of TAVI23. In addition to the above-mentioned valves, which have been implanted in humans, there are numerous other devices that are under development. These are: the self-expanding Colibri valve (Colibri Heart valves, LLC, Broomfield, CO, USA), which is expected to be implanted in humans this year, the AorTx valve (Hansen Medical Inc, Mountain View, CA, USA), the Trinity Flexx valve (Transcatheter Technologies, Regensburg, Germany), the UCL TAV (University College of London, London, UK), which in contrast to the others has polymeric leaflets, the Vanguard™ II valve (ValveXchange Inc, Greenwood Village, CO, USA), in which the leaflets are exchangeable when the valve requires replacement, the Optimum TAV (Thubrikar Aortic Valve Inc, Rapid City, SD, USA) and the HLT valve (Heart Leaflet Technologies Inc, Maple Grove, MN, USA), which was implanted in humans in 2009, and after which it was decided to redesign it. Recently, Edwards Lifesciences (USA) announced the development of two additional valves: the Centera and the SAPIEN III. The first is a self-expanding valve that can be delivered through the transfemoral approach. It has bovine pericardial tissue leaflets and its deployment is performed with the use of a 14 Fr motorised delivery system. The SAPIEN III valve also has bovine pericardial leaflets and has been designed for transfemoral and transapical delivery. Advantages of the valve are its low profile (it requires a 14 Fr sheath for transfemoral and an 18 Fr for transapical delivery) and the fact that it includes a customised sealing cuff that may reduce/prevent the risk of paravalvular leaks. Both new Edwards devices have entered the first-in-man stage.
EMBOLIC PROTECTION DEVICES
Cerebrovascular adverse events are well-recognised complications of TAVI. The PARTNER trial reported a 6.7% event rate in high-risk patients who cannot undergo surgical aortic valve replacement (SAVR) and a 5.5% event rate in patients with increased comorbidities who however were deemed suitable for SAVR24,25. In addition, magnetic resonance imaging (MRI) studies have demonstrated subclinical post-procedural embolic event rates in up to 91% of patients26. Mechanical cerebral protection devices have been developed to address this issue (Figure 3).

Figure 3. Available mechanical cerebral protection devices: A) the Claret CE Pro (Claret Medical, Inc, Santa Rosa, CA, USA); B) the Shimon Embolic Filter (SHEF) (SMT R&D, Herzliya, Israel); C) the Embrella embolic protector (Edwards Lifesciences, Irvine, CA, USA).
CLARET CE PRO
The Claret CE Pro system (Claret Medical, Inc., Santa Rosa, CA, USA) is designed to filter the blood in the brachiocephalic and left common carotid artery. It has two filters: the proximal and the distal. The proximal filter is attached to a long catheter and consists of a polyurethane filter sewn onto a radiopaque nitinol frame that can be apposed and seals the wall of the brachiocephalic artery. The distal filter is deployed after the proximal filter and covers the left common carotid artery. The system is compatible with a 6 Fr sheath and can be introduced through the right radial or brachial artery. The effectiveness of this device was recently tested in a small feasibility study that included 40 patients who underwent TAVI with the CoreValve®. Both generation systems were used of which the second had a better safety and efficacy profile. The overall success rate of the two devices was 88% for the proximal filter and 73% for the distal filter. No cerebrovascular events were noted during the procedure. However, there were two minor and one major stroke within the first month post operation. Debris was macroscopically detected in 54% of the filters that were successfully deployed27. Although this study demonstrated the feasibility of the Claret CE Pro system, further evaluation is needed in the context of randomised control trials to evaluate its effectiveness in protecting patients from cerebrovascular events.
SHIMON EMBOLIC FILTER™ (SHEF)
In contrast to Claret CE Pro, which functions as a debris capture system, the Shimon Embolic Filter (SHEF) (SMT R&D, Herzliya, Israel) operates as a debris deflector. It consists of a heparinised mesh that is attached to a nitinol frame, which has stabilisers on its upper side that help to fix the device in the aortic arch. The SHEF device also protects the left subclavian artery and can stay in position for days or even for months. The system is deployed through the femoral artery and requires a 9 Fr catheter. This device was tested in the SMT FIM feasibility study in which 15 patients received the device during TAVI. According to the results reported at TCT 2011 meeting there were no clinically evident neurological events during the procedure but two patients suffered a stroke at day four and five post TAVI while the MRI scan showed a 50% reduction in new cerebral lesions compared to the control group28. Currently the DEFLECT I study is underway, which will incorporate head MRI before and after TAVI to provide further evidence about the effectiveness of SHEF.
EMBRELLA EMBOLIC DEFLECTOR
Embrella embolic protector (Edwards Lifesciences, Irvine, CA, USA) is the third system designed to cover the cranial vessels during TAVI. This includes an oval-shaped nitinol frame that is covered with a porous membrane (100 μm). The device is an embolic deflector that is deployed in the aortic arch to protect the brachiocephalic and the left carotid artery, it is compatible with a 6 Fr sheath and is delivered through the right radial or brachial artery. The Embrella embolic deflector was successfully deployed for the first time in January 2010 and tested in a small feasibility study, which was recently reported in the TCT 201129. In this study, 18 patients were included in whom the devices effectively covered both the left carotid and the brachiocephalic arteries. Post-procedure MRI in 15 subjects demonstrated a reduced number of new cerebral lesions per patient (3.2 lesions/patient), results that compared favourably to those reported in studies that did not use a cerebral protection device26,30. Based on these encouraging data a feasibility study has been designed, named PROTAVI, which will investigate the effectiveness of Embrella in a larger set of patients.
OTHER UPCOMING CLINICAL TRIALS
According to the available data, TAVI treatment is a valuable option in patients with symptomatic aortic stenosis and a (very) high operative risk who are deemed inoperable24,25. However, as the interventionists become more familiar with the procedure and are able to appreciate its capabilities and potentialities it is inevitable that they will shift their attention to populations with lower interventional risk. Thus today, numerous trials are underway or are expected to start aiming to investigate the effectiveness of TAVI in younger and lower-risk patients.
COREVALVE VS. SAVR-DENMARK TRIAL
This randomised single-blind control trial was designed to compare TAVI using the CoreValve with surgical aortic valve replacement (SAVR) in patients >70 years old. The study started in 2008 and is expected to finish in 2013. Within this period, 280 patients will be randomised on a 1:1 basis and they will be followed up for five years. The primary and secondary endpoints of this trial are illustrated in Table 1.
MEDTRONIC COREVALVE® U.S. PIVOTAL TRIAL
This trial started at the end of 2010 and has two cohorts: the “extreme risk” and the “high risk” cohort. The “extreme risk” cohort will investigate the safety and efficacy of the CoreValve in the treatment of symptomatic patients deemed inoperable for aortic valve surgery. Initially it was designed to randomise patients on a 2:1 basis to TAVI and optimal medical treatment but the protocol changed at the beginning of the trial, which was modified to a single-arm study that would compare its outcomes to those reported in other trials (including the PARTNER cohort B).
The “high risk” cohort aims to demonstrate that TAVI with the CoreValve is not inferior to SAVR and will include 790 patients, with a predicted risk of operative mortality ≥15%, who will be randomised on a 1:1 basis to TAVI with the CoreValve and SAVR. The patients will be followed up for five years; the primary and secondary endpoints are illustrated in Table 1.
PARTNER II TRIAL
The PARTNER II trial has been recently started and is divided in two cohorts: A and B. In cohort A, 2,000 patients, with a Society of Thoracic Surgeons risk score of ≥4, will be randomised on a 1:1 basis to TAVI with the Edwards SAPIEN XT valve and SAVR. Patients with coronary artery disease (CAD) and a SYNTAX score of <33 will be included in the study and there will be a sub-stratification according to the presence of CAD (Table 1). Patients with CAD will be randomised to TAVI plus percutaneous coronary intervention and to SAVR plus coronary bypass grafting on a 1:1 basis. A detailed neurological evaluation before and post procedure will be included and a frailty assessment will be performed in each subject, which will provide data for a frailty substudy. All patients will be followed up for at least two years. Cohort B will randomise 500 inoperable patients to TAVI with the Edwards SAPIEN THV and Edwards SAPIEN XT on a 1:1 basis and will compare the safety and efficacy of the two devices (Table 1). Twenty percent of the studied population will undergo a detailed neurological assessment pre and post procedure to identify new cerebrovascular events. In addition, this study will include a comparison of the two devices in inoperable patients without vascular access who will undergo transapical TAVI. The enrolment started in 2011 and the study is expected to finish in 2018.
SURTAVI TRIAL
The SURgical and Transcatheter Aortic Valve Implantation (SURTAVI) trial is a non-inferiority prospective randomised trial that aims to recruit 1,800 intermediate risk patients who will be randomised on a 1:1 basis to TAVI with the CoreValve and SAVR. The trial has already started and approximately 75 centres from Europe and the USA are participating. Patients with CAD will not be excluded from the study as long as they have a SYNTAX score of <23. The primary objective is to compare the efficacy of TAVI and SAVR in intermediate risk patients. However, this study also has numerous secondary objectives including the detection of differences in the quality of life and health economics between the two groups. A detailed neurological assessment before and post procedure is also included that will allow a more accurate estimation of the occurrence of cerebrovascular adverse events after TAVI treatment. The duration of this trial is approximately seven years. The enrolment flowchart is illustrated in Figure 4 and the primary and secondary endpoints in Table 1.
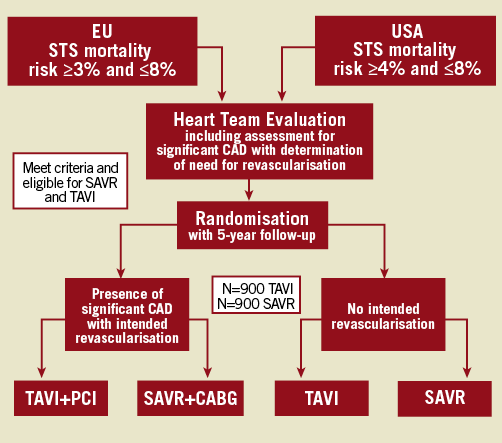
Figure 4. Enrolment flowchart for the SURTAVI trial. EU: European Union; USA: United States of America; STS: Society of Thoracic Surgeons; CAD: coronary artery disease; SAVR: surgical aortic valve replacement; TAVI: transcatheter aortic valve implantation; PCI: percutaneous coronary intervention; CABG: coronary artery bypass grafting
Conclusions
TAVI is maturing as a less invasive approach for treating patients with severe aortic stenosis. The current evidence from large registries and randomised control trials has demonstrated its feasibility and efficacy and drawn the attention of industry and physicians. Several new devices have been developed that are easy to use, have more adjustment tools and reduce the risk of complications. These advances as well as the fact that the interventionists have become more familiar with the procedure have increased the applications of TAVI and nowadays there are several ongoing trials that aim to identify the type of patients who would benefit from this treatment. It is apparent that we are at the dawn of an exciting era for percutaneous valvular interventions. The future is expected to be more prosperous as new developments and data from ongoing trials will provide the background to expand TAVI applications and establish their position in a broader spectrum of patients.
Acknowledgements
The authors would like to express their appreciation to cvPipeline (http://www.cvpipeline.com/) for generously providing up-to-date information about the new developments in the field of TAVI. C.V. Bourantas would also like to acknowledge the funding support provided by the Hellenic Heart Foundation (ELIKAR) Athens, Greece.
Conflict of interest statement
The authors have no conflict of interest to declare.
