
Abstract
Transcatheter aortic valve implantation (TAVI) has rapidly evolved and changed the landscape of structural interventional cardiology. Advances in transcatheter heart valve (THV) prostheses and TAVI-enabling devices have simplified the procedure, reduced the risk of complications, improved short- and long-term outcomes and broadened the applications of TAVI, not only in challenging patients and complex anatomies but also in intermediate-risk or even in low-risk patients, where surgical valve replacement constitutes an effective and well-established therapy. In this review article, we provide an overview of the developments in TAVI devices which have played a vital role in TAVI evolution: we describe the prostheses that failed to reach clinical practice, we present the characteristics of the first valves that were tested in the clinical arena, we summarise the evidence from the first studies that highlighted the potential but also the limitations of TAVI, and we present the advanced next-generation THV prostheses that have an improved performance and safety profile.
Abbreviations
CE: Conformité Européenne
FIM: first-in-man
PET: polyethylene terephthalate
PHV: percutaneous heart valve
PVR: paravalvular regurgitation
SAVR: surgical aortic valve replacement
TAVI: transcatheter aortic valve implantation
THV: transcatheter heart valve
Introduction
Transcatheter aortic valve implantation (TAVI) was introduced at the beginning of this century and revolutionised the treatment of aortic valve disease. The first clinical studies that examined the safety and efficacy of TAVI provided proof of the concept and attracted the interest of the scientific community and industry who intensified their efforts and designed new devices that overcame the limitations of the first prototypes and broadened the clinical applications of TAVI1.
Over the last 10 years there has been a revolution in TAVI technology: several valves have been introduced that simplified the procedure, reduced complications and improved procedural outcomes. These advances have changed the landscape in structural interventions and rendered TAVI an effective treatment for symptomatic patients with aortic valve disease. In this review article we provide an overview of the developments in valve devices: we describe the first prototypes and the evidence from the first clinical studies (Supplementary Table 1), highlight their limitations, describe the modifications that were made in updated revisions and present the unique qualities of the next generation of TAVI systems (Supplementary Table 2). Finally, we describe the characteristics of the devices that have not been used in the clinical arena, aiming to highlight the challenges, diversity and competition in the field.
Evolving TAVI devices
PERCUTANEOUS HEART VALVE
The percutaneous heart valve (PHV) was the first transcatheter aortic valve to be implanted in humans. The first prototype consisted of three bovine pericardial leaflets mounted on a stainless steel balloon-expandable stent and implanted from the femoral vein using a 24 Fr sheath. In the I-REVIVE and RECAST trials – the first clinical studies that examined the efficacy of TAVI – a revised version of the PHV was used: the modified valve had equine pericardial leaflets and was implanted using both the retrograde and antegrade approach. The RECAST study demonstrated a success rate of 75% and a high incidence of paravalvular regurgitation (PVR, ≥grade 2 in 63% of the patients) (Supplementary Table 1). To address this limitation, the Cribier-Edwards valve (Edwards Lifesciences, Irvine, CA, USA) was introduced. This valve incorporated a sewn fabric cuff covering the left ventricular portion of the prosthesis: the valve could be deployed through the femoral artery using a 22-24 Fr sheath and dedicated deflection catheter, or from the transapical approach using a 33 Fr sheath2,3.
An update of this prototype, the Edwards SAPIEN transcatheter heart valve (THV; Edwards Lifesciences) was introduced in 2006 and became the first TAVI system to acquire Conformité Européenne (CE) mark approval (Figure 1). The prosthesis had leaflets made from bovine pericardium and a sealing cuff made of polyethylene terephthalate (PET). It was used in the PARTNER 1 cohort A and B studies, which assessed the efficacy of TAVI against medical therapy in inoperable patients, and against surgical aortic valve replacement (SAVR) in high-risk patients, respectively4,5. These studies established TAVI as an effective treatment in these populations but also revealed some significant limitations of the technology. In particular, the high incidence of cerebrovascular events, vascular complications and moderate to severe PVR noted at 30-day follow-up led to the refinement of the prosthesis and the introduction of the SAPIEN XT™ valve (Edwards Lifesciences). This revision had a cobalt-chromium frame with thinner struts and an open-cell design, while the leaflets of the prosthesis were semi-closed when the valve was in its natural position (to reduce the time and pressure difference required for diastolic closure). The device was implanted from the transfemoral approach using the NovaFlex (Edwards Lifesciences) delivery system that allowed balloon mounting of the valve in the aorta. These modifications resulted in a lower crossing profile (18-19 Fr) of the NovaFlex system compared to the RetroFlex (22-24 Fr) delivery system (Edwards Lifesciences) used for deployment of the SAPIEN THV valve. A prospective study comparing the two devices showed that these modifications resulted in a 67% reduction of the major vascular complications rate using the SAPIEN XT valve (11.1% vs. 33.3%, p=0.004)6.
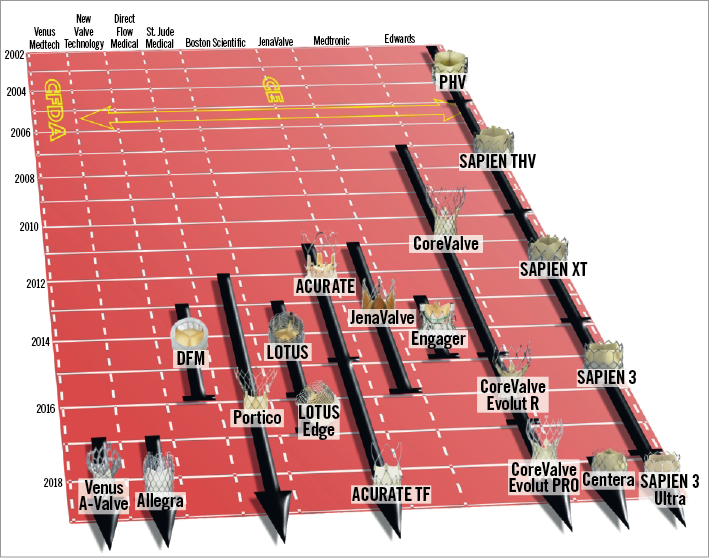
Figure 1. Evolution of TAVI systems with CE mark or China Food and Drug Administration approval. Prostheses are presented according to the date of regulatory approval. Images were obtained and modified with permission from Bourantas et al19.
The SAPIEN XT device was used in the PARTNER 2 randomised controlled study comparing clinical outcomes following TAVI and SAVR in intermediate-risk patients with severe aortic stenosis7. The investigators demonstrated no difference between the two groups in the primary endpoint (all-cause mortality or disabling stroke: 19.3% vs. 21.1%, p=0.25) at two-year follow-up. However, the incidence of major vascular complications (8.6% vs. 5.5%, p=0.006) and moderate/severe PVR was higher in the TAVI arm (8.0% vs. 0.6%, p<0.001), while acute kidney injury (3.8% vs. 6.2%, p=0.02), life-threatening or disabling bleeding (17.3% vs. 47.0%, p<0.001) and new-onset atrial fibrillation (11.3% vs. 27.3%, p<0.001) were increased in the SAVR arm. To reduce the incidence of PVR and to improve safety, Edwards Lifesciences introduced the SAPIEN 3 valve which had a smaller crimping profile, a longer cobalt-chromium open-cell stent frame, and an additional PET skirt. Transfemoral deployment is achieved using the 14-16 Fr eSheath™ (outer diameter for the 20 mm, 23 mm and 26 mm prosthesis 6.0 mm, and 6.7 mm for the 29 mm prosthesis) and the Commander Delivery System™ (both Edwards Lifesciences) which has a distal flex point to facilitate coaxial positioning. Transapical implantation is performed using an 18-21 Fr sheath and the Certitude™ delivery system (Edwards Lifesciences). Device safety and efficacy were tested in the SAPIEN 3 study that included 1,077 intermediate-risk patients. A low incidence of death and disabling stroke was noted at one-year follow-up, while vascular complications, moderate/severe PVR, and the need for reintervention arose in 6.1%, 2% and 1%, respectively (Supplementary Table 1). The incidence of pacemaker implantation at one-year follow-up was 12.4%, 2.5% higher than the incidence of pacemaker implantation reported in the SAPIEN XT arm of the PARTNER 2 study.
A propensity-matched analysis against patients who underwent SAVR in PARTNER 2 showed that TAVI with the SAPIEN 3 valve was associated with better outcomes than SAVR. The SAPIEN 3 valve is currently being assessed in the randomised PARTNER 3 study (NCT02675114) comparing the safety and efficacy of TAVI and SAVR in 1,328 low-risk patients with severe aortic stenosis.
Edwards has recently introduced an advanced revision of the SAPIEN 3, the SAPIEN 3 Ultra prosthesis. The valve has a 40% taller PET skirt with an up to 50% greater contact surface area with the native valve anatomy, which is expected to reduce the incidence of PVR further. The device is implanted through a 14 Fr delivery system. It was recently tested in the SAPIEN 3 Ultra CE mark study (NCT03471065) and acquired CE mark approval in November 2018.
COREVALVE
The CoreValve® (Medtronic, Minneapolis, MN, USA) was the first self-expanding valve introduced into clinical practice for the treatment of severe aortic stenosis. The device incorporates porcine pericardial leaflets mounted on a nitinol frame and a pericardial skirt to reduce the risk of PVR; its implantation is performed only via the antegrade approach using the 18 Fr AccuTrak™ delivery system (Medtronic). The safety and efficacy of the prosthesis were examined in the CoreValve Extreme Risk Pivotal Trial that included 506 inoperable patients with symptomatic severe aortic stenosis. The procedural success rate was 77.6%, while the incidence of all-cause mortality, stroke, major vascular complications, pacemaker implantation and of moderate to severe PVR was 24.3%, 7.0%, 8.4%, 21.6% and 4.2%, respectively, at one-year follow-up (Supplementary Table 1).
The CoreValve high-risk study was the first randomised trial that compared SAVR and TAVI with a self-expanding prosthesis. It included 795 high-risk patients who were followed up for one year (Supplementary Table 1). The study reported a lower incidence of death (14.2% vs. 19.1%, p=0.04), acute kidney injury (15.6% vs. 6.0%, p<0.001) and new-onset atrial fibrillation (32.7% vs. 15.9%, p<0.001) in the TAVI arm and a higher incidence of major vascular complications (6.2% vs. 2.0%, p=0.004), pacemaker implantation (22.3% vs. 11.3%, p<0.001) and moderate/severe PVR (7.0% vs. 1.3%, p<0.01) in this group. There were no differences between groups in the incidence of stroke (8.8% vs. 12.6%, p=0.100).
In an attempt to improve the safety profile, Medtronic introduced the CoreValve Evolut™ R system with a shorter nitinol frame to fit better in angulated anatomies and increased radial force to optimise annular sealing. The pericardial skirt maintained its original height and extended within the inflow tract to minimise the risk of PVR, while the porcine pericardial tissue leaflets were treated with alpha-amino oleic acid to reduce leaflet calcification and enhance device durability. Device implantation is performed using the EnVeo R delivery system (Medtronic) through a lower profile (inner diameter: 18 Fr, outer diameter 6.0 mm for the 23 mm, 26 mm and 29 mm prosthesis and 6.7 mm for 34 mm) in-line sheath; the EnVeo R system enables controlled device deployment and re-sheathing, repositioning and re-deployment if necessary. The CoreValve and Evolut R prostheses were used in the recently published SURTAVI study that compared TAVI and SAVR in 1,660 intermediate-risk patients with symptomatic aortic stenosis8. There were no differences between the two groups in the primary endpoint (all-cause mortality or disabling stroke: 12.6% vs. 14.0%, p for non-inferiority >0.999) at two-year follow-up; however, the incidence of reintervention (2.8% vs. 0.7%) and hospitalisation for aortic valve-related disease (13.2% vs. 9.7%) was higher in the TAVI arm, relating to the higher rate of moderate/severe PVR in this group (3.4% vs. 0.7%).
The safety and efficacy of the Evolut R prosthesis have also been tested in the FORWARD study that included 1,038 patients. In this study the mortality rate was 1.9% at 30-day follow-up while the incidence of stroke, pacemaker implantation and moderate/severe PVR was 2.8%, 19.7% and 2.0%, respectively (Supplementary Table 1).
To reduce the risk of PVR, Medtronic has recently introduced an updated revision, the CoreValve Evolut™ PRO that incorporates an additional external porcine pericardial tissue wrap covering the first 1.5 cells of the device which has a 20 Fr crimping profile (outer diameter of the sheath 6.7 mm). The Medtronic Evolut PRO US clinical study that included 60 patients with severe symptomatic aortic stenosis reported one death and one disabling stroke at one-month follow-up while none of the studied patients had moderate/severe PVR (Supplementary Table 1). These promising results provided the substrate for the conduct of the FORWARD PRO study that aims to examine the long-term (five-year) performance of the prosthesis in 600 high-risk patients (NCT03417011) and of the Medtronic Evolut Transcatheter Aortic Valve Replacement in Low Risk Patients study (NCT02701283) that aims to compare TAVI and SAVR in 1,200 low-risk patients with severe aortic stenosis.
JENAVALVE
JenaValve™ (JenaValve, Munich, Germany) is a self-expanding prosthesis that incorporates three porcine tissue leaflets attached to a nitinol frame, a porcine pericardial skirt to reduce the incidence of PVR, and three feelers which are designed to embrace the native valve leaflets, providing tactile feedback and coaxial implantation. This special configuration renders the device effective for the treatment of aortic stenosis and aortic regurgitation9. Implantation is performed via a transapical approach using a 32 Fr delivery system. Despite robust evidence supporting the safety and efficacy of the JenaValve in the clinical arena, the prosthesis was removed from the market in June 2016 because of the limited use of the transapical access (Supplementary Table 1).
Recently, the Everdur™ Plus prosthesis (JenaValve) was introduced to allow transfemoral implantation. Similar to the JenaValve, the device incorporates three feelers that allow clipping of the prosthesis to the native leaflets. The first-in-man (FIM) study raised initial concerns about the safety of the delivery system that has subsequently been re-designed. Today, device implantation is performed using the Coronatix transfemoral delivery catheter (JenaValve) through a 19 Fr sheath. The safety and efficacy of this prosthesis are currently being tested in the Everdur CE Mark Trial-AS study.
ACURATE VALVE
The ACURATE TA™ prosthesis (Symetis SA, Ecublens, Switzerland) is a self-expanding valve that consists of porcine tissue leaflets mounted on a nitinol frame and has three arches that provide better stability during implantation. The lower/distal edge of the device forms a crown that is designed to provide axial fixation and is covered by a PET sealing skirt to reduce PVR. The device is implanted via the transapical approach using a sheathless 28 Fr system. The safety and efficacy of the prosthesis were tested in the SAVI registry that included 250 patients and reported 6.8% mortality, 2.8% incidence of stroke and no significant PVR at 30-day follow-up (Supplementary Table 1).
To enable transfemoral implantation, Symetis modified the first revision and introduced the ACURATE neo™ prosthesis. This prototype has more flexible stabilisation arches, an upper crown for supra-annular anchoring and more stable positioning, and a pericardial skirt that is incorporated into the stent body and lower crown to reduce PVR. Implantation is performed using a flexible 18 Fr delivery system. The SAVI-TF registry examined the safety and efficacy of the ACURATE neo in 1,000 high-risk patients and reported favourable one-year results (Supplementary Table 1).
In 2017, Boston Scientific acquired Symetis SA and recently introduced the ACURATE neo2™ advanced sealing system with a modified outer skirt to reduce the incidence of PVR further. The safety and performance of the device have recently been tested in the ACURATE neo AS TF CE Mark study that included 120 patients and reported promising 30-day outcomes (Supplementary Table 1).
LOTUS VALVE
The Sadra® Lotus™ (Boston Scientific, Marlborough, MA, USA) was the first fully retrievable and repositionable transcatheter prosthesis. The first revision had three bovine pericardial leaflets mounted in a braided nitinol frame, an outer sealing membrane (Adaptive Seal™; Boston Scientific) to reduce PVR, a radiopaque marker to facilitate correct positioning, and three locking mechanisms to allow initial evaluation and repositioning if required before release. The prosthesis was initially delivered using a 21 Fr system, but an updated version was introduced in 2009 that allowed controlled mechanical expansion of the valve through an 18-20 Fr sheath.
REPRISE II was the first large study that examined the safety and efficacy of the second Lotus device in 120 high-risk patients. It demonstrated a high incidence of stroke (5.9%) and pacemaker implantation (28.6%) at 30-day follow-up10. Stroke rates were lower (3.0% and 4.8%) in the larger RESPOND and REPRISE 3 studies (n=996, n=912) while the incidence of pacemaker implantation remained high (34.6% and 35.5%, respectively) in these reports11,12. To minimise the injury caused in the conduction system during valve implantation, Boston Scientific designed the LOTUS Edge™ valve that incorporates the Depth Guard™ deployment technology which is expected to minimise the depth of valve implantation and thus reduce the need for pacemaker implantation (Walters L. First report of clinical outcomes with the next-generation Lotus Edge valve system: results from the Lotus edge feasibility trial. Presented at ACC 2017, Washington, DC, USA, 17-19 March 2017). The device acquired CE mark approval in 2016 but was removed from global commercial use in 2017 due to reports of premature release of the pin connecting the valve and delivery system.
A fourth-generation Lotus valve has recently been designed and is anticipated to undergo FIM studies in the near future.
HLT VALVE
The HLT™ valve (HLT, Inc., Maple Grove, MN, USA) consists of a glutaraldehyde cross-linked tricuspid porcine pericardial tissue valve, an elastic nitinol frame, a nitinol mesh that supports the valve and a braided polyester liner to minimise PVR. The device is uniquely inverted before implantation to anchor the native valve and is retrievable and repositionable (Figure 2A). The FIM study conducted in 2009 encountered a high complication rate and was stopped because of safety concerns. The valve system was redesigned and the updated Meridian® valve and the 18 Fr Pathfinder® delivery system (both HLT, Inc.) were successfully tested in the RADIANT US/Canada study (Supplementary Table 1). The RADIANT CE mark trial has recently commenced and will include 200 intermediate/high-risk patients to examine device safety and efficacy.
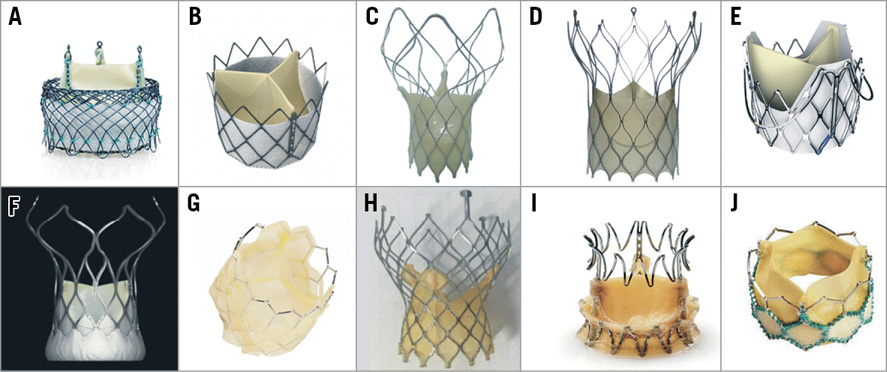
Figure 2. Examples of prostheses that have been implanted in humans but do not yet have CE mark approval. A) Meridian; B) Inovare; C) Hydra*; D) Biovalve#; E) J-Valve; F) VitaFlow; G) Colibri Heart Valve; H) Venibri; I) Trinity; J) MyVal. Reprinted with permission from Bourantas et al19, and Mercanti et al20. *Reprinted from Srimahachota et al21 with permission from Europa Digital & Publishing. #Reprinted from Treede et al22 with permission from Europa Digital & Publishing.
Valve devices that have been evaluated in the clinical arena
PORTICO VALVE
The Portico™ valve, designed by St. Jude Medical (St. Paul, MN, USA) and bought by Abbott (Abbott Vascular, Santa Clara, CA, USA), consists of three leaflets manufactured from bovine pericardial tissue attached to a nitinol stent frame whose inflow is covered by a bovine pericardial tissue skirt. The prosthesis has a large-cell design to reduce the risk of PVR and ease coronary access and is implanted via the antegrade approach using an 18-19 Fr delivery system.
The Portico TF EU and Portico 1 studies examined the safety and efficacy of the device and demonstrated a 30-day mortality of 2.7-3.6%, a stroke rate of 2.4-3.2%, and an incidence of major vascular complications of 5.5-7.2%, while the incidence of moderate PVR was 3.9-5.7%13,14. The valve has had CE mark approval since 2012.
INOVARE VALVE
The Inovare® valve (Braile Biomedica, São José do Rio Preto, Brazil) (Figure 2B) is a prosthesis that has been developed in a partnership by industry, academia and government agencies to cover the needs of Brazil15. The valve is balloon-expandable and consists of a cobalt-chromium frame with three radiopaque markers for optimal device deployment and three leaflets created by a single sheet of bovine pericardium. Implantation can be performed via the transfemoral or transapical approach using a 24 Fr delivery system16. A recent report that examined the performance of the device in 90 high-risk patients undergoing transapical TAVI demonstrated favourable procedural results, while small case series have also demonstrated the feasibility of transfemoral delivery15,16. The valve is used in Brazil but does not have CE mark approval.
CENTERA VALVE
CENTERA (Edwards Lifesciences) is a self-expanding valve that incorporates three bovine pericardial tissue leaflets mounted on a nitinol frame and a PET skirt to minimise the risk of PVR. In contrast to other self-expanding devices, the prosthesis has a short stent frame that facilitates correct valve positioning. Deployment is achieved via transfemoral approach using a motorised delivery system and an expandable 14 Fr sheath.
A recent study that examined the safety and efficacy of the CENTERA in 198 high-risk patients reported an overall 30-day mortality rate of 1.0%, and an incidence of pacemaker implantation of 4.9%, while stroke and moderate/severe PVR was reported in 4.0% and 0.6%, respectively (Supplementary Table 1). The ongoing ExCEED randomised study (NCT03517436) aims to compare outcomes following TAVI with the CENTERA and SAVR in 1,000 intermediate-risk patients with severe symptomatic aortic stenosis.
VENUS A-VALVE
The Venus A-Valve® (Venus Medtech, Hangzhou, China) is a self-expanding prosthesis that incorporates three porcine pericardial leaflets which are attached supra-annularly to a nitinol frame. The device has three radiopaque markers to facilitate correct positioning and is deployed via the transfemoral approach using an 18-20 Fr delivery system that allows retrieval and repositioning if required. Safety and effectiveness have recently been tested in the Venus A trial that included 101 intermediate- or high-risk patients with severe bicuspid or tricuspid aortic stenosis (Supplementary Table 1). In this study the incidence of procedural complications was similar to previous TAVI studies, while the incidence of moderate/severe PVR was high (9.2%); however, these findings should be interpreted with caution since echocardiographic analysis was not performed by a core lab. This device is the first to acquire approval for use in China from the China Food and Drug Administration (Figure 1).
HYDRA VALVE
The Hydra valve (Vascular Innovations Co., Ltd, Nonthaburi, Thailand) (Figure 2C) is a self-expanding prosthesis that incorporates bovine pericardial leaflets attached to a nitinol stent frame. The device is implanted via transfemoral approach using an 18 Fr delivery system and is retrievable and repositionable. A FIM study has recently been completed and the reported results are promising (Supplementary Table 1).
BIOVALVE
The Biovalve (Biotronik AG, Bülach, Switzerland) (Figure 2D) is a self-expanding valve system consisting of three leaflets and a skirt made from porcine pericardium that are attached to a nitinol frame which has increased inflow radial force to ensure optimal device anchoring and minimal PVR. Implantation is performed via the transfemoral approach using an 18 Fr delivery system.
The prosthesis has recently been tested in the BIOVALVE-I study, a small feasibility study that included 13 high-risk subjects (Supplementary Table 1). The larger BIOVALVE-II study (NCT02249000) has recently commenced and aims to assess the safety and efficacy of the device in 73 high-risk patients.
J-VALVE
The J-Valve™ (Jiecheng Medical Technology Co., Ltd., Suzhou, China) (Figure 2E) system consists of three porcine leaflets attached to a self-expanding nitinol frame. The device has three U-shaped graspers that facilitate coaxial positioning and axial and radial fixation. The valve is deployed via the transapical or the transfemoral approach using a 27 Fr or 18 Fr delivery system, respectively17. FIM studies have shown that the prosthesis is safe and effective for the treatment of both aortic stenosis and regurgitation (Supplementary Table 1).
VITAFLOW VALVE
The VitaFlow valve (MicroPort® Medical, Shanghai, China) (Figure 2F) is a self-expanding prosthesis that has three bovine pericardial leaflets mounted on a self-expanding nitinol frame. The device has an inner and outer skirt to minimise PVR and is implanted via the transfemoral approach using a 16-18 Fr motorised delivery system. Valve performance was recently validated in a pre-market clinical study that included 110 inoperable patients with one-year clinical outcomes comparable to those reported in other valve studies (Supplementary Table 1). To simplify the procedure and optimise device deployment, MicroPort has recently designed the VitaFlow™ delivery system that incorporates a motorised handle to enable valve retrieval and repositioning. The updated system is currently being tested in the VitaFlow™ II Transcatheter Aortic Valve System Study (NCT03575039) that aims to recruit 178 inoperable patients with severe aortic stenosis. The device is anticipated to gain China Food and Drug Administration approval in the near future.
ALLEGRA NVT VALVE
The Allegra NVT valve (New Valve Technology, Hechingen, Germany) consists of three bovine pericardial tissue leaflets attached to a self-expanding frame with a closed-cell diamond-shaped configuration. The device incorporates six radiopaque gold markers to facilitate correct placement, while its ventricular inflow is covered with a bovine pericardial skirt to minimise the risk of PVR. Transfemoral implantation is performed using an 18 Fr sheath and the Permaflow delivery system (New Valve Technology) that allows device recapturing and repositioning if required. The FIM study included 21 inoperable patients and showed 95.2% procedural success and favourable clinical and haemodynamic results at 30-day follow-up (Supplementary Table 1). The recently commenced FOLLOW study (NCT03613246) will report the short-term and midterm clinical outcomes in 200 high-risk patients undergoing TAVI with this prosthesis.
Other TAVI devices
Apart from the above-mentioned TAVI prostheses that are used in the clinical setting, there are several other devices that had clinical applications in the past but have now been removed from the market, as well as prostheses that are currently undergoing FIM studies and prototypes that were introduced in the past but have not reached the clinical arena (Supplementary Appendix 1, Figure 2G-Figure 2J, Figure 3).
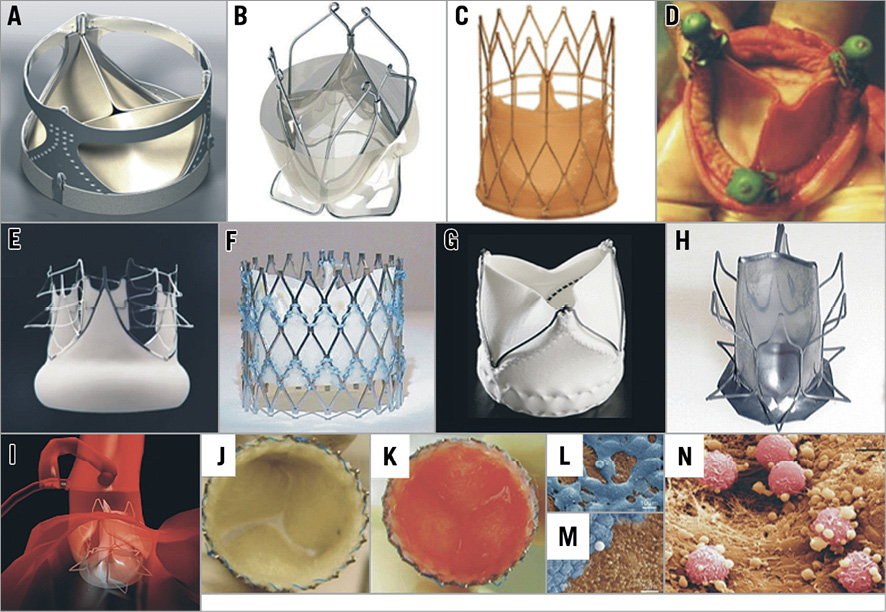
Figure 3. Prototypes assessed in animal studies but not yet implanted in humans. A) AorTx valve; B) Triskele; C) FoldaValve*; D) Vanguard II valve; E) Optimum TAV; F) Sat TAVI system; G) Xeltis prosthesis#; H) & I) PercValv (the device consists of a thin film eNitinol membrane and has a monolithic design); and J) DJ valve (the prosthesis leaflets consist of a non-woven poly-glycolic acid mesh seeded with bone marrow mononuclear cells using fibrin as cell carrier [K]). Electron microscopy images obtained from a valve explanted from a baboon model four weeks following implantation demonstrate endothelial cells (L), thrombocytes (M) and leukocytes (N) attached to the valve surface. Reprinted with permission from Bourantas et al19 and Bourantas et al1. *Reprinted from Kheradvar et al23 with permission from Europa Digital & Publishing. # Reprinted from Myazaki et al24 with permission from Europa Digital & Publishing.
Conclusions
TAVI has matured over the last 10 years and is now regarded as an established treatment for patients with severe aortic stenosis. Cumulative data have provided robust evidence about the durability of the TAVI prostheses18. Currently, several ongoing studies aim to establish TAVI as the first treatment option for low-risk patients suffering from severe aortic stenosis. This evolution would not have been possible without revolutionary changes in TAVI devices that have reduced the crimping profile, improved durability and haemodynamic performance, decreased rates of PVR and simplified the procedure. In parallel, several cheaper devices have been designed to address the needs of developing countries and reduce the cost of the procedure. Credit for this evolution should be given to the first pioneers who introduced the concept, the clinicians who supported the technology and contributed with novel ideas and patient data, the research that introduced revolutionary and futurist approaches, and the industry that appreciated the potential of TAVI, invested in this endeavour, spread the technology and created a highly competitive market that rewarded creativity and innovations. The strong dynamics between companies working in the field led to occasional conflicts, closure, merger or collaboration of different companies, but more importantly led to technical developments that changed the field and opened new horizons in structural intervention. Future research is expected to focus not only on the identification of patients who will benefit from this therapy and the development of easy-to-use prostheses that will reduce the incidence of vascular complications, PVR, and pacemaker implantation, but also on the identification of ideal valve characteristics for the treatment of specific challenging anatomies associated with increased periprocedural risk and adverse prognosis.
Guest Editor
This paper was guest edited by Alec Vahanian, MD, PhD; Department of Cardiology, Hôpital Bichat-Claude Bernard, and University Paris VII, Paris, France.
Acknowledgements
The authors would like to acknowledge the support of Hans Jonker who contributed to the editing and preparation of the Figures.
Conflict of interest statement
A. Baumbach has received institutional research support from Abbott Vascular. L. Søndergaard has received institutional research grants and consultant fees from Abbott Vascular, Boston Scientific, Edwards Lifesciences, Medtronic and Symetis. B. Prendergast has received fees and unrestricted research grants from Edwards Lifesciences. S. Kennon has received consultation fees from Edwards Lifesciences. M. Mullen has received institutional research support from Edwards Lifesciences. P. Serruys has received personal fees from Medtronic. The other authors have no conflicts of interest to declare. The Guest Editor is a consultant for Edwards Lifesciences.
The complete list of references can be found in the online version of this paper.
Supplementary data
Supplementary Appendix 1. Other TAVI devices.
Supplementary Table 1. Studies assessing the safety and efficacy of TAVI devices implanted in humans.
Supplementary Table 2. Morphological characteristics of CE mark-approved devices and prostheses under evaluation in current clinical studies.
To read the full content of this article, please download the PDF.
