
Abstract
Aims: Percutaneous mitral valve repair has become an alternative to conventional surgery in patients suffering primary mitral regurgitation (MR) with a contraindication to surgery and could benefit patients at high surgical risk. The aim of the MITRA-HR study is to raise the level of evidence supporting the use of the MitraClip device in primary MR patients with a predefined high risk for surgery.
Methods and results: MITRA-HR is a prospective, multicentre, randomised study designed to compare mitral valve repair using the MitraClip with conventional surgery in patients with severe primary mitral regurgitation at high risk for surgery. The surgical risk is defined by age, Society of Thoracic Surgeons (STS) risk estimate score, frailty, major organ system dysfunction, and procedure-specific impediments. The study will enrol 330 patients randomised between conventional surgery and MitraClip with a 1:1 ratio. The composite primary endpoint includes all-cause mortality, unplanned rehospitalisation for cardiovascular reasons, and mitral valve reintervention at 12 months. The main secondary safety endpoint is a major adverse event composite assessment evaluated 30 days after the procedure.
Conclusions: The randomised MITRA-HR study is designed to provide additional supportive evidence of non-inferiority in efficacy and superiority in safety for percutaneous mitral valve repair using the MitraClip compared to conventional surgery in high surgical risk patients.
Introduction
Mitral regurgitation (MR) is one of the most common heart valve disorders in Western countries1. MR is classified as either primary MR (a structural abnormality of the mitral valve apparatus) or secondary MR (a disease of the left ventricle or the left atrium which interferes with the function of the mitral valve apparatus without structural abnormality). Primary MR covers all aetiologies in which intrinsic lesions affect one or several components of the mitral valve apparatus. The gold standard therapeutic management of severe primary MR is surgery to repair or replace the mitral valve2,3. However, 50% of patients are not referred for surgery because of age or presence of comorbidities, which increase the surgical risk3,4.Percutaneous repair of the mitral valve with MitraClip® (Abbott Vascular, Santa Clara, CA, USA) performed according to the edge-to-edge procedure has become an alternative to conventional mitral valve surgery. The randomised Endovascular Valve Edge-to-Edge Repair Study (EVEREST II) comparing this technique with surgery in patients with primary or secondary MR eligible for mitral valve surgery following the guidelines showed better safety for the percutaneous approach with similar functional results but more residual MR and need for reintervention than for surgery5,6. Current guidelines2,3 agree that the percutaneous edge-to-edge procedure may be considered in patients with symptomatic severe primary MR who fulfil the echocardiographic criteria of eligibility, are judged inoperable or at high surgical risk by a Heart Team, and have a life expectancy greater than one year (recommendation class IIb, level of evidence C).
However, no randomised study has compared the MitraClip procedure with surgery in primary MR patients at high surgical risk. We therefore set out to conduct such a study assuming that, for this population, repair with the MitraClip could be non-inferior in efficacy and superior in safety compared to conventional surgery.
Methods
STUDY DESIGN AND OBJECTIVES
MITRA-HR is a prospective, multicentre, randomised, open-label study of the MitraClip system for the treatment of severe primary MR in patients who present a high risk as assessed by the Heart Team but no contraindication for surgery.
The primary objective is to show that endovascular treatment with the MitraClip in this population is not inferior to surgical treatment by assessing the occurrence at 12 months of a composite endpoint including all-cause mortality, unplanned rehospitalisation for cardiovascular reasons, and mitral valve reintervention. The main secondary objective is to compare the two strategies in terms of safety by assessing the occurrence of a major adverse event composite endpoint at 30 days.
The study will be performed according to the principles of the Declaration of Helsinki, the guidelines for Good Clinical Practice of the International Conference on Harmonization (ICH-E6), the French national guidelines for good clinical practice as regards biomedical research on medicinal products for human use, the national laws and regulations, and the European Clinical Trials Directives 2001/20/EC and 2005/28/EC. All participants will provide written informed consent prior to enrolment in the study, which has been approved by the Ethics Committee (Comité de Protection des Personnes Est III, Nancy, France; decision dated 7 November 2017).
The trial is registered at www.clinicaltrials.gov (identifier: NCT03271762).
STUDY DEVICE AND PROCEDURES
The MitraClip system received CE mark approval in Europe in 2008 and U.S. Food and Drug Administration approval in 2013. A newer device (MitraClip NT®, 2016 CE mark approval in Europe) with an easily steerable guide catheter will be used in this study. The implantation technique has been described previously7.
Percutaneous repair of the mitral valve with the MitraClip device will be compared with conventional surgery, which favours mitral valve repair whenever technically feasible over valve replacement, as recommended in current guidelines2,3.
STUDY POPULATION
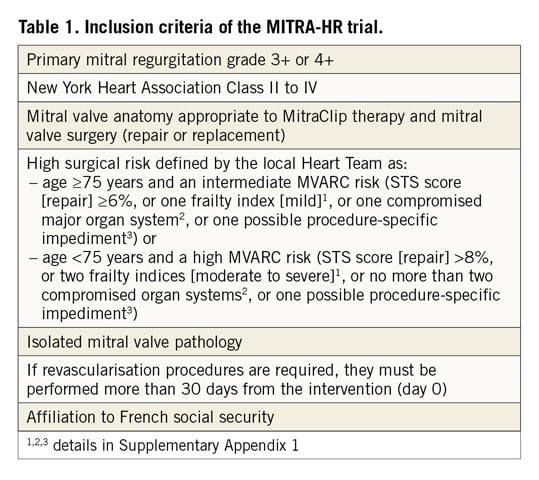
Eligible patients are those who have severe primary MR as defined in the guidelines3. They should fulfil the requirements for anatomical repair with the MitraClip and for mitral valve surgery with high surgical risk. The surgical risk, which is central to defining the target population, is assessed by the Heart Team, composed of, at least, a clinician cardiologist, an interventional cardiologist, a cardiac surgeon, an echocardiographer and an anaesthesio-logist. It is defined by the age of the patient and the composite risk assessment provided by the Mitral Valve Academic Research Consortium (MVARC), which combines the Society of Thoracic Surgeons (STS) risk estimate score for mitral valve repair, frailty indices, major organ system dysfunction criteria, and procedure-specific impediments8,9. Inclusion criteria are listed in Table 1. The definitions of frailty indices, major organ system compromise and procedure-specific impediments are shown in Supplementary Appendix 1. Exclusion criteria are listed in Supplementary Table 1.
SUBJECT SCREENING, ENROLMENT, AND RANDOMISATION
The recruitment of the patients is performed in selected French centres with experience in cardiac valve surgery and interventional cardiology. All patients are screened for eligibility (Figure 1) after signing the informed consent form (within 45 days prior to the intervention).
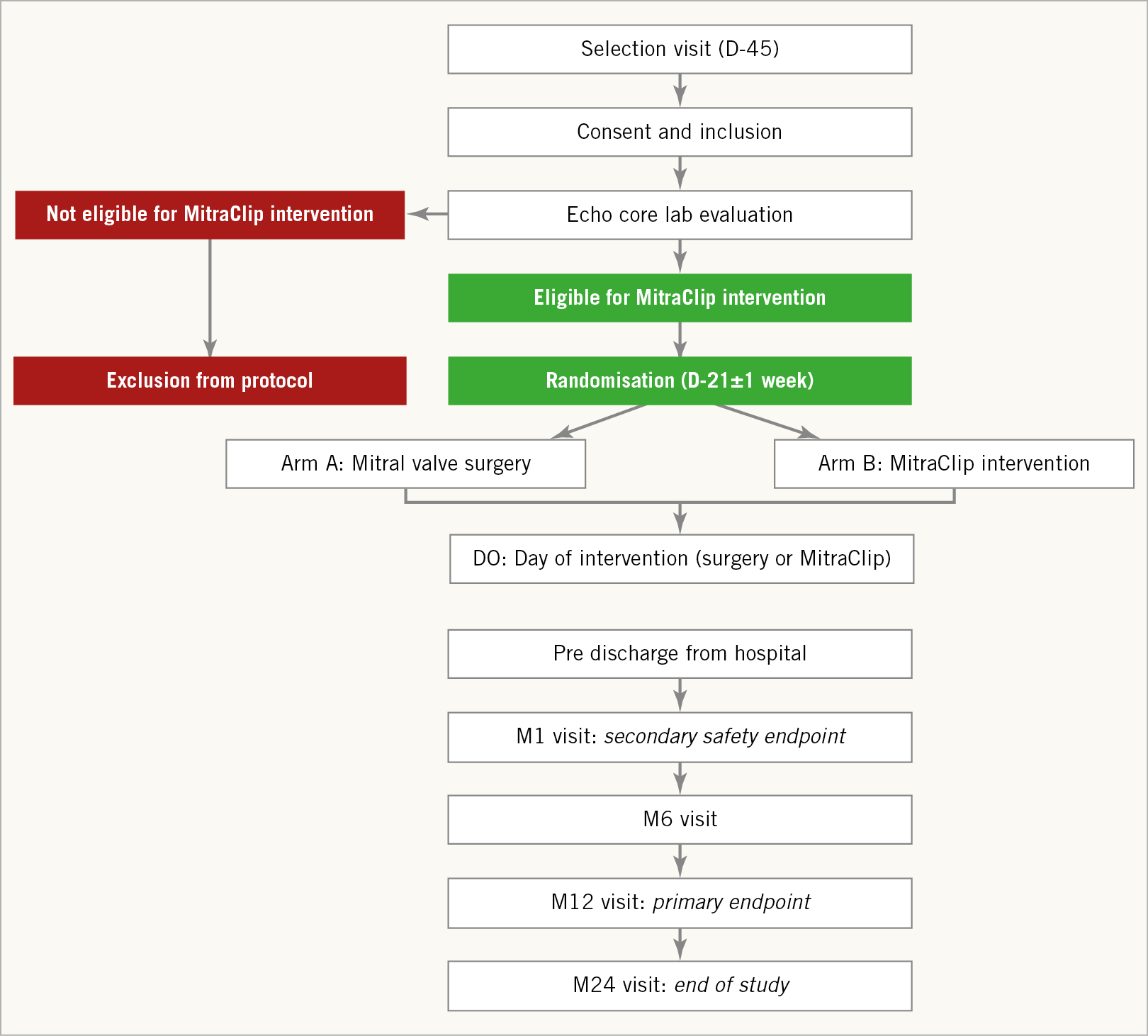
Figure 1. MITRA-HR study design. D0: day zero (intervention); D-21 and D-45: day 21 and day 45 before intervention; M1, M6, M12, and M24: months one, six, twelve and twenty-four after intervention
The Heart Team confirms the absence of contraindication for mitral valve surgery and the presence of a high surgical risk.
Transthoracic echocardiographic (TTE) and transoesophageal echocardiographic (TOE) data are reviewed by a centralised core laboratory (Rennes CHU), which validates the patients’ eligibility for a MitraClip intervention based on previously published echocardiographic selection criteria10 (Supplementary Table 2).
All enrolled patients have to be eligible for surgery and for MitraClip intervention.
The patients’ characteristics and clinical data at inclusion are collected using an electronic case report form.
Eligible patients are randomised 21±7 days before the intervention using a secure website. The randomisation ratio for the MitraClip arm and the conventional surgery arm is 1:1.
PATIENT FOLLOW-UP: ASSESSMENTS AND ENDPOINTS
Clinical visits at study centres are performed 30 days, 6 months, 12 months, 18 months and 24 months after the procedure.
The primary endpoint is a composite of all-cause mortality, unplanned rehospitalisation for cardiovascular reasons and mitral valve reintervention at 12 months following the procedure.
The main secondary safety endpoint, evaluated 30 days after the procedure, is a major adverse event composite derived from MVARC recommendations8,9 (Supplementary Table 3). The overall rates of surgery-related serious adverse events and serious adverse device effects are also assessed at 6, 12, and 24 months.
Other secondary endpoints evaluated at 6, 12 and 24 months include all-cause mortality, cardiovascular mortality, unplanned heart failure rehospitalisation, unplanned rehospitalisation for cardiovascular reasons, mitral valve reintervention, change in NYHA class, quality of life, biological parameters (B-type natriuretic peptide [BNP] or N-terminal pro-B-type natriuretic peptide [NT-proBNP] levels) and renal function. Additional secondary endpoints include residual MR grade, left and right cardiac chamber remodelling and function at 30 days and 12 months, and change in six-minute walk test at six and 12 months.
ENDPOINT ADJUDICATION
An independent safety and efficacy committee will evaluate all potential clinical events related to the primary endpoint as well as major cardiovascular events. This adjudication committee will reduce physician bias and allow a greater degree of control over the criteria used for assessing patient outcome. The adjudicators will be blinded to patient treatment allocation and their decisions will be used for all statistical analyses. The adjudication committee will be solicited once a year for the adverse events review.
STUDY SAFETY MONITORING
As the study is interventional with minimal risk and constraint, an independent data safety monitoring board will not be necessary.
STATISTICAL ANALYSIS
The study is designed to demonstrate the non-inferiority of MitraClip to surgery on the primary composite endpoint at 12 months.
In the high-risk MR population enrolled in the study, the 12-month rate of occurrence of the composite primary endpoint is expected to be about 20% (approximately 8-10% mortality5,11,12,13, 8-10% rehospitalisation for cardiovascular reasons14, and 2% mitral valve reintervention5). The clinically relevant non-inferiority margin is set at 13%, the power at 80% and the alpha risk at 2.5%. With these assumptions, 330 patients (165 per group) are needed to show the non-inferiority of MitraClip to surgery on the primary endpoint, accounting for loss of follow-up (10%). With a recruitment estimate of six patients per year and per centre, the study population can be recruited within 36 months.
The primary analysis is carried out on the per-protocol population. Missing data are imputed as worst case scenarios. A secondary intention-to-treat analysis will be performed15.
Demographic and clinical data are recorded for each patient. For binary variables, counts and percentages are calculated. For continuous variables, means, standard deviations, minima-maxima are presented. For time-to-event variables, survival curves are constructed using the Kaplan-Meier method.
The composite primary endpoint including all-cause mortality, unplanned rehospitalisation for cardiovascular reasons, and mitral valve reintervention is calculated for each arm and the percentage difference between the two groups is presented with a two-sided 95% confidence interval. The lower limit of the 95% confidence interval is compared with the non-inferiority limit (13%). Confidence intervals are estimated by mixed linear models to take into account stratification by centre. Multivariate mixed models are used to study the effect of specific risk factors.
The Student’s t-test is used for comparative analyses of continuous variables and Fisher’s exact test for categorical variables. The Kaplan-Meier estimates are compared with the log-rank test. The global and specific survival rates are determined between the randomisation date and the event date. For patients who die before the final follow-up examination or for patients lost to follow-up, the status of the last follow-up examination is recorded. Mixed models are used to study the evolution of parameters on follow-up.
A subgroup analysis is performed for patients achieving a residual MR ≤1+.
With regard to the cost-effectiveness analysis, 95% confidence intervals for differential costs and quality-adjusted life years (QALYs) and for the incremental cost-effectiveness ratio are estimated using the non-parametric bootstrap estimation procedure.
No interim analysis for efficacy based on the primary endpoint is planned.
Analyses will be performed using SPSS Statistics for Windows, Version 19 (IBM Corp., Armonk, NY, USA) and SAS for Windows, version 9.4 (SAS Institute Inc., Cary, NC, USA).
TRIAL ORGANISATION
The MITRA-HR study is supported by the Nantes University Hospital (CHU de Nantes) and monitored by the Sponsor Department of CHU de Nantes. The study principal investigator is Professor Patrice Guérin. Dr Ousama Al Habash and Dr Nicolas Piriou are additional scientific coordinators. Data management is performed by Mrs Emilie Leblanc (Promotion Department, CHU de Nantes). Statistical analyses are performed by Mrs Béatrice Guyomarch from the Institut du Thorax (CHU de Nantes). The health-economic analysis will be carried out by Dr Philippe Teissier and Dr Valéry-Pierre Riche (Innovation and Partnership Department, CHU de Nantes/Université de Nantes). Members of the steering and adjudication committees are listed in Supplementary Appendix 2.
The centres participating in the MITRA-HR study (Supplementary Appendix 3) have been selected for their experience in valvular cardiac surgery and interventional cardiology. Only centres performing at least 50 cases of mitral surgery per year and having performed at least 10 MitraClip procedures (including at least two procedures with the new MitraClip NT) can participate.
Results
The first patient was enrolled in March 2018. No results are available as the study is currently recruiting.
Discussion
Surgical mitral valve repair or replacement is the treatment of choice for severe primary MR as confirmed by the latest European Society of Cardiology and American Heart Association/American College of Cardiology guidelines2,3. Although transcatheter repair has emerged as an alternative for treating primary MR in patients presenting a contraindication to surgery, it has not been specifically investigated in a randomised study in vulnerable patients with a recognised high risk for surgery.
The evidence for the efficacy and safety of the MitraClip is drawn from numerous clinical trials and registries5,6,16,17,18,19,20, with follow-up data available for up to five years after the procedure. The EVEREST II trial is the only randomised study that has compared MitraClip percutaneous repair with surgery5. The study included patients with severe MR (74% primary MR) and no contraindication for surgery (mean STS mortality risk: 5±4%). At 12 months, the occurrence rates of the primary efficacy endpoint (freedom from death, from surgery for mitral valve dysfunction, and from grade 3+ or 4+ MR) were 55% in the percutaneous repair group and 73% in the surgery group (p=0.007). The difference was driven by increased rates of surgery for mitral valve dysfunction after percutaneous repair (20% versus 2%, respectively; p<0.001). It has to be noted that this trial was performed during the early learning curve of the MitraClip procedure, when success rates were lower than those observed in more recent registries (77% acute procedural success rate versus 91 to 97%)5,16,19,20. On the other hand, percutaneous treatment was associated with a reduction in the rate of major adverse events at 30 days (p<0.001). The final five-year results of the EVEREST II trial supported the superiority of surgery in reducing MR but also confirmed the long-term safety of the MitraClip device6. Interestingly, in subgroup analyses of the EVEREST II trial, surgery was not superior to percutaneous repair in older patients (≥70 years) and patients with left ventricular dysfunction (left ventricular ejection fraction [LVEF] <60%)5,6.
Several registries and single-arm studies16,19,21,22,23 have suggested that patients with a high risk for surgery may benefit from a less invasive percutaneous mitral valve repair. According to the ACCESS-EU registry, most patients currently receiving a MitraClip device in Europe have a higher surgical risk profile than those evaluated in the EVEREST II trial and are mainly treated for secondary MR16. They are older, more symptomatic, and have more comorbidities. Yet, in this high-risk patient population, the MitraClip procedure is safe and effective, with sustained improvements in quality of life and functional status at 12 months. In the EVEREST II High Risk Study, MitraClip reduced MR in a majority of patients deemed at high risk for surgery, and was associated with improvement in clinical symptoms and one-year survival, when compared with a matched population receiving standard care (medical therapy or conventional surgery)22. A subsequent study similarly showed that, despite a higher logistic EuroSCORE, high surgical risk patients with symptomatic severe MR treated with MitraClip have similar survival rates when compared with matched surgically treated patients, with both populations showing a survival benefit compared with matched medically treated patients23. Taken together, these studies suggest that percutaneous mitral valve repair is a favourable alternative for patients with severe primary MR presenting a high risk for surgery. However, the relevance of these studies is hampered by their observational design with inherent limitations due to uncontrolled confounding factors. This is of particular importance in elderly high-risk patients with comorbidities who represent a particularly heterogeneous population, in which there is a particular likelihood of confounding factors.
For the purposes of the randomised MITRA-HR study, high risk for surgery is defined by the age of the patient and a composite risk assessment provided by MVARC, which combines STS risk estimate score, frailty, major organ system dysfunction, and procedure-specific impediments8,9. For this population with a predefined high surgical risk but no contraindication to surgery, our hypothesis is that repair with the MitraClip could be as effective and safe as conventional surgery. Defining the non-inferiority margin at 13% was a critical issue in the design process of the study. The results of the EVEREST II trial were criticised for the large non-inferiority margins used (31% and 25% for the per-protocol and “comparison of strategy” analyses, respectively)24. Our composite endpoint was constructed from hard outcomes (mortality, mitral valve reintervention) and soft outcomes (unplanned rehospitalisation for cardiovascular reasons). Any difference in hard outcomes may be considered as clinically meaningful and warrants the choice of a narrow, conservative margin. In contrast, a reduced efficacy and therefore a wider margin may be acceptable if the outcome is a soft outcome or a composite including a soft outcome.
Data concerning the occurrence of the components of the primary endpoint in the population defined in our study are limited. There are no data on variability and size effect from historical data and/or randomised trials. On the basis of the available data, we have anticipated the occurrence of the primary endpoint in the surgical group to be about 20%. Hence, MitraClip therapy will be considered non-inferior to surgery as long as the primary endpoint is lower than the upper limit of the 95% confidence interval, i.e., around 33%.
Limitations
Site-based variability in patient selection can be a limitation, particularly in the surgical risk assessment. However, MVARC clinical trial design principles still agree that the local Heart Team is more appropriate to determine the relative surgical risk and operability of a patient, rather than a central eligibility committee8. To ensure that a single site does not induce bias because of a too discordant surgical risk assessment compared to the other sites, sensitivity analyses could be considered if single site outcomes in either arm are outliers.
The authors are fully aware that the non-inferiority margin of 13% for an expected primary outcome of 20% in the surgical group can lead to a limited value in the interpretation of the primary endpoint. However, pre-specified analysis of each individual component of the primary endpoint balanced with 30-day safety analysis should allow a pertinent clinical interpretation of the MITRA-HR trial data.
Conclusions
The randomised MITRA-HR study is designed to provide additional supportive evidence of non-inferiority in efficacy and superiority in safety for percutaneous mitral valve repair using the MitraClip device compared to conventional surgery in high surgical risk patients.
|
Impact on daily practice For patients with severe primary MR presenting a high surgical risk but no contraindication to surgery, there is an unmet medical need for a safer alternative to valve surgery; however, the level of evidence supporting percutaneous mitral valve repair is low. The MITRA-HR study will provide important evidence as to whether the MitraClip may be considered as an alternative to surgery for this high-risk patient group. |
Acknowledgements
The authors thank Imen Fellah, Valéry-Pierre Riche, Philippe Teissier, Anne Chiffoleau, Marine Renard, Caroline Postnikoff and Alexandra Poinas for their contribution to this work.
Funding
This study is supported by a grant from the French Ministry of Health (PHRC 2017) and a partnership with Abbott Vascular that provides devices at no cost.
Conflict of interest statement
N. Piriou has served as consultant for Abbott Vascular and has received fees. P. Guérin has served as consultant for Abbott Vascular and has received fees; he has also received speaker fees from General Electric. T. Le Tourneau and J.C. Roussel have received a grant from St. Jude-Abbott, outside the submitted work. J.F. Obadia has served as consultant for Abbott Vascular, Edwards, Medtronic, Landanger, Delacroix Chevalier and has received fees. A. Vahanian has received speaker honoraria from Abbott Vascular, Edwards Lifesciences, and consultancy fees from Mitral Tech. B. Iung has received speaker fees from Edwards Lifesciences, Boehringer Ingelheim and Novartis. E. Donal, J.N. Trochu and O. Al Habash have received no financial support from Abbott Vascular. The other authors have no conflicts of interest to declare.
