Abstract
Background: Stroke remains a feared complication associated with transcatheter aortic valve implantation (TAVI). Embolic cerebral injury occurs in the majority of TAVI cases and can lead to cognitive dysfunction.
Aims: The PROTEMBO C Trial evaluated the safety and performance of the ProtEmbo Cerebral Protection System in TAVI patients.
Methods: Forty-one patients were enrolled in this single-arm study conducted at 8 European centres. The primary safety endpoint was the rate of VARC 2-defined major adverse cardiac and cerebrovascular events (MACCE) at 30 days; the primary performance endpoint was the composite rate of technical success versus performance goals (PG). Secondary endpoints included brain diffusion-weighted magnetic resonance imaging (DW-MRI), new lesion volume, and the rate of death or all strokes compared to historical data.
Results: Thirty-seven of 41 enrolled patients underwent TAVI with the ProtEmbo device (intention-to-treat [ITT] population). Both primary endpoints were met. MACCE at 30 days was 8.1% (upper limit of the 95% confidence interval [CI]: 21.3% vs PG 25%; p=0.009), and technical success was 94.6% (lower limit of the 95% CI: 82.3% vs PG 75%; p=0.003). New DW-MRI lesion volumes with ProtEmbo were smaller than in historical data, and 87% of patients completing MRI follow-up had no single lesion >150 mm3. There was 1 stroke in a patient in whom the device was removed prematurely before TAVI completion.
Conclusions: The PROTEMBO C Trial met its primary safety and performance endpoints compared to prespecified historical PGs. Patients had smaller brain lesion volumes on DW-MRI compared to prior series and no larger single lesions. These results warrant further evaluation of the ProtEmbo in a larger randomised controlled trial (RCT).
Introduction
Transcatheter aortic valve implantation (TAVI) is an established alternative to surgical aortic valve replacement for treatment of severe symptomatic aortic valve stenosis12, with more than 276,000 procedures performed in the United States between 2011 and 20193. Despite the broad adoption of TAVI4, and the procedure now being performed in low-risk patients5678, stroke remains a major complication associated with significant mortality and morbidity79. The incidence of stroke at 30 days ranges from 3.3%1 to 12%10 and is associated with a 6-fold increase in mortality11, reduced quality of life12, and significant economic burden2.
Stroke complicating TAVI primarily results from embolic debris released during TAVI, with embolic material consisting of cholesterol particles, air, atherosclerotic plaque material, thrombus, and calcified valve material13141516. Beyond acutely symptomatic stroke, upward of 94% of TAVI patients have evidence of embolic ischaemic brain injury on diffusion-weighted magnetic resonance images (DW-MRI), which has been associated with long-term cognitive dysfunction and motor deficits11718192021.
Cerebral embolic protection devices (CEPDs) have been developed to prevent new neuroembolic cerebral events and so-called silent infarctions122. In the United States, CEPDs are currently available in less than one-third of TAVI centres and used in only 13% of TAVI procedures23. The low penetration of CEPDs into clinical practice reflects the ongoing debate (and lack of evidence) regarding the effectiveness of CEPDs in reducing stroke. Recent randomised clinical trials evaluating CEPDs have demonstrated safety without convincing evidence of benefit in reducing stroke or total lesion volume on magnetic resonance imaging (MRI)923, whereas propensity-adjusted real-world data have suggested neurological and mortality benefits24. Results of a definitive large-scale randomised trial evaluating a single CEPD are expected in late 202225.
The ProtEmbo Cerebral Protection System (Protembis GmbH) is a novel CEPD with a 60 µm pore size designed to protect all 3 cerebral vessels. It is the only left radial access device currently under development. The ProtEmbo was shown to be safe and feasible in the first-in-human PROTEMBO SF Trial (ClinicalTrials.gov: NCT03325283)26. The objective of the PROTEMBO C Trial (ClinicalTrials.gov: NCT04618718) was to evaluate the safety and performance of the ProtEmbo for embolic protection during TAVI.
Methods
Trial design and oversight
The PROTEMBO C Trial is an international, multicentre, single-arm, non-inferiority study designed to evaluate the safety and performance of the ProtEmbo System in patients with severe symptomatic native aortic valve stenosis undergoing TAVI (Supplementary Appendix 1). The study was performed in compliance with the Declaration of Helsinki and was approved by the ethics committee of each contributing centre. Each patient provided informed consent. Data collection and monitoring were performed independently by a clinical research organisation (MAXIS Medical). An independent data and safety monitoring board of 2 interventional cardiologists, 1 cardiac surgeon, and 1 neurologist oversaw the safe conduct of the study and adjudicated all clinical events.
Patient population
Patients with severe symptomatic calcified native aortic valve stenosis who met approved indications for TAVI with commercially available transcatheter aortic valves by transfemoral access were eligible for the study. Patients were excluded if TAVI was planned using access other than transfemoral access or had any of the following: a previously implanted heart valve; evidence of acute myocardial infarction, transient ischaemic attacks, or cerebrovascular accidents within the prior 6 months; blood dyscrasias; contraindications to aspirin, heparin, antiplatelet/anticoagulant therapy, or device materials; renal or hepatic insufficiency; participation in other trials; or any other planned permanent cardiac implant within 30 days of the index procedure. Other exclusion criteria were neurological impairments, a contraindication to MRI, excessive vascular tortuosity or severe peripheral arterial disease, an abnormal aortic arch angulation or anatomy or an inner diameter of the aortic arch <25 mm. Patients meeting all eligibility criteria who signed informed consent and who received a baseline MRI were considered enrolled in the study (Supplementary Appendix 1- Supplementary Appendix 11).
ProtEmbo device and trial procedures
The ProtEmbo device is a temporary, single-use, intra-aortic embolic deflection filter used as an adjunct device during TAVI that is the only available device that can be positioned through a 6 Fr left radial access sheath. The ProtEmbo is inserted at the beginning of the procedure, after administration of heparin with an adequate activated clotting time (ACT) level above 250 seconds, prior to the TAVI device, and removed following the completion of the TAVI procedure. The device consists of (1) a heparin-coated, 60 µm pore size mesh (currently the smallest pore size of CEPDs), (2) a self-expanding nitinol frame that measures 38×70 mm when expanded to ensure sufficient coverage with radiopaque markers for fluoroscopic visualisation and precise device placement, and (3) a delivery unit. The device is delivered unexpanded and deployed by unsheathing the self-expanding filter to cover the orifice of all 3 cerebral vessels (brachiocephalic trunk, left common carotid, and left subclavian arteries) (Central illustration). A handle provides a simple user interface for preparation, delivery, deployment, and removal of the device. The device is loaded into a commercially available delivery catheter and placed into the aortic arch using a commercially available guiding sheath via the left radial or brachial artery. TAVI procedures were performed according to institutional standards. Clinical evaluations included assessments at baseline, post-procedure, before discharge, and at 30 days.
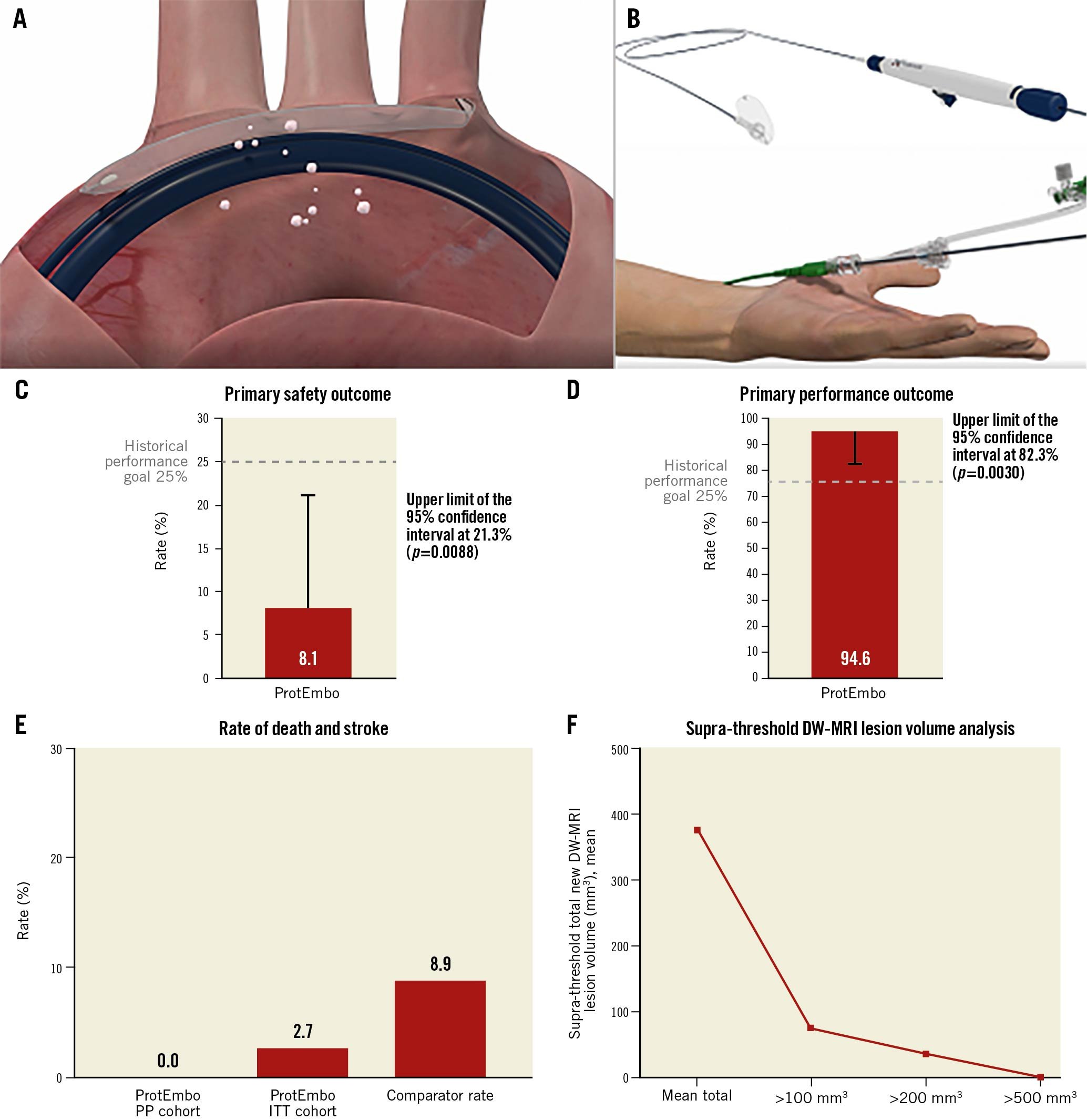
Central illustration. ProtEmbo and summary of results. A). The ProtEmbo deployed in the aortic arch showing the delivery over radial access and coverage of 3 main cerebral vessels with the TAVI catheter, deflecting embolic debris. B) Illustration of the functional parts of the ProtEmbo without handle. The ProtEmbo met (C) the primary safety outcome and (D) the primary performance outcome compared with historical performance goals. E) The secondary efficacy analysis (death or all stroke at 30 days) compared to 9.9% of the SENTINEL trial control arm (N=111)30 and 7.0% in the REFLECT II trial control arm (N=57)2. F) The supra-threshold lesion volume analysis for the ProtEmbo. DW-MRI: diffusion-weighted magnetic resonance imaging; ITT: intention-to-treat; PP: per protocol; TAVI: transcatheter aortic valve implantation
Brain DW-MRI scans were acquired at baseline (at least 5 days after any diagnostic cardiac catheterisation) and 2-7 days after TAVI. The timing and acquisition of 1.5 Tesla DW-MRI was standardised across all study centres based on a detailed acquisition protocol. All MRIs were analysed by an independent core laboratory (Buffalo Neuroimaging Analysis Center) using established methods for assessments of ischaemic lesions.
Endpoints and outcome measures
The primary safety endpoint was major adverse cardiac and cerebrovascular events (MACCE) at 30 days: defined as a composite of all-cause mortality, all stroke, life-threatening or disabling bleeding, major vascular complications in the access vessels or aorta, or acute kidney injury (stage 2 or 3), all according to the Valve Academic Research Consortium-2 (VARC-2) criteria27. Secondary safety endpoints included stroke severity, quantified acutely using the National Institutes of Health Stroke Scale (NIHSS) score (NeuroARC)28, and the occurrence of other serious adverse events reported up to 30 days.
The primary performance endpoint was the rate of technical success: defined as the ability to safely deliver, deploy, and remove the device; the ability to secure and stabilise the position of the device throughout the procedure; and to deflect embolic material, defined by coverage of the 3 cerebral vessels without impeding blood flow. Adequate stability and coverage of the 3 cerebral vessels in the aortic arch was assessed by means of angiographic review by the investigator at each site. Mild-to-moderate interactions (no device displacement) were deemed acceptable, while severe interaction (displacement of the device to the ascending or descending aorta) was considered technical failure.
Secondary efficacy endpoints included a composite of death or VARC-2-defined strokes at 72 hours and 30 days compared to historical data and the median total new lesion volume assessed by brain DW-MRI at 2 to 7 days compared to historical data. The total new lesion volume was defined as the sum of all diffusion-positive new cerebral lesions in the post-procedural DW-MRI relative to the pre-TAVI DW-MRI.
Histological analysis of the ProtEmbo device was conducted by an independent core laboratory (CVPath Institute) to assess the haemocompatibility of the ProtEmbo device surface and to characterise debris captured by the device.
Statistical analysis
The study prespecified 3 populations for analysis. The safety cohort comprised patients with signed informed consent and completed baseline DW-MRI assessment. The intention-to-treat (ITT) cohort included patients in the safety cohort who met the eligibility criteria with an attempt to use the ProtEmbo (device passed through the skin). The per protocol (PP) cohort included patients in the ITT cohort who received treatment with the ProtEmbo device in accordance with the protocol and completed both MRIs (baseline and follow-up at 2-7 days). The primary safety and performance endpoints were assessed in the ITT cohort and the secondary efficacy endpoints were assessed in the PP cohort.
The primary safety endpoint of 30-day MACCE with the ProtEmbo was compared with a performance goal (PG) of 25% derived from historic data. A sample size of 60 patients would provide 85% power to reject the null hypothesis (the upper limit of the 95% confidence interval [CI] of 30-day MACCE with the ProtEmbo using the Wilson Method was less than the PG) assuming a 30-day MACCE with the ProtEmbo of 10% and a 1-sided alpha=0.02529.
The primary performance endpoint for the ProtEmbo was compared to an historic PG of 75%. A sample size of 42 patients would provide 85% power to reject the null hypothesis (the lower limit of the 95% CI with the ProtEmbo using the Wilson method was greater than the PG) assuming a success rate for the ProtEmbo of 89% and a 1-sided alpha=0.02529. Study success requires both primary endpoints to be met.
The secondary efficacy MRI endpoints are summarised using descriptive statistics28, using median values when not normally distributed30. A multi-threshold, lesion-wise analysis for each patient investigated the supra-threshold new lesion volumes above incremental thresholds from >100 to >1000 mm3, where lesions below the respective thresholds were excluded from the mean and compared with historical data.
The study was terminated early (with enrolment of 41/60 planned patients) after meeting the primary safety and performance endpoints. Bootstrapping using Stata (StataCorp) was conducted and reviewed by the independent data and safety monitoring board to ensure that the conclusions of the trial were fully justified for both primary endpoints. The bootstrapping analysis for each primary endpoint was performed on 5,000 simulated 60-patient samples and was used to generate the lower 95% CI of the performance endpoint and the upper 95% CI of the safety endpoint, since these are the key determinants of the non-inferiority test.
Results
Patient and procedural characteristics
A total of 56 patients were screened for the procedure, and 41 patients were enrolled in the study (safety cohort); 37 patients underwent TAVI using the ProtEmbo device (ITT cohort), of which 31 were treated according to protocol and underwent DW-MRI (PP cohort). Patients were 46% male with a mean Society of Thoracic Surgeons (STS) score of 2.81±1.36%, and 22% were treated with self-expanding valves (Table 1, Table 2).
TAVI was successful in all 37 patients in the ITT cohort using either the Medtronic Evolut R or PRO, Edwards SAPIEN 3, or Meril Myval; 1 patient required 2 TAVIs due to a residual large paravalvular leak. The use of the ProtEmbo was attempted in 37 patients and was successful in 94.6% (35/37). The average time for device deployment was 4.5±4.9 minutes, the average device dwell time in the blood stream was 30.2±13.4 minutes (range 16 to 79 minutes), the amount of mean additional contrast used was 5.9±16.7 mL (the majority of patients [27/37] received no additional contrast for the use of the ProtEmbo), and the additional fluoroscopy time was 4.3±4.5 minutes (Table 2).
Safety outcome
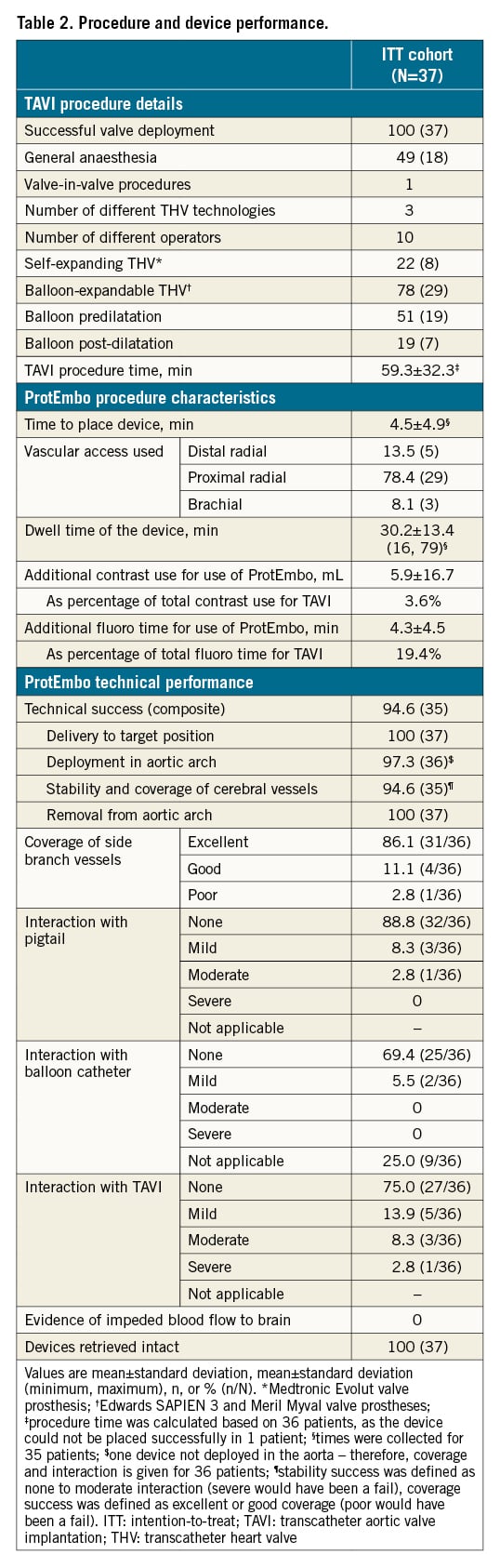
MACCE at 30 days in the ITT cohort was 8.1% (3/37), meeting the predefined PG for the safety endpoint (upper limit 95% CI: 21.3% vs PG 25%; p=0.009) (Central illustration). There were no deaths and no device-related adverse events in this trial. Table 3 summarises MACCE events, which included a cardiac tamponade unrelated to the ProtEmbo, a thalamic cerebral infarct that developed 12 hours after the TAVI procedure in which the ProtEmbo was retrieved prematurely due to interaction between the TAVI catheter and the ProtEmbo, and an acute kidney injury (stage 3) requiring dialysis in a patient with chronic renal insufficiency prior to TAVI. There was no significant worsening of NIHSS in any of the patients with complete follow-up. Vascular access site-related complications in the radial or brachial artery occurred in 5 of the 37 patients enrolled in the ITT cohort, of which 2 were asymptomatic and 8.1% (3 of the 37 patients) were symptomatic (for 2 events, conservative management was sufficient, and for 1 event, the bleeding was stopped by applying a peripheral balloon).
Performance outcome
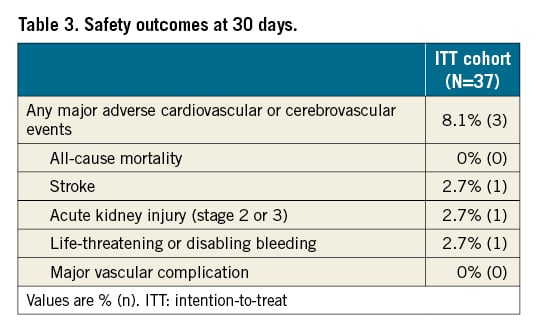
Technical success was achieved in 94.6% (35 of 37) patients in the ITT cohort, which met the prespecified endpoint compared with the PG (lower 95% CI: 82.3% vs PG 75%; p=0.003) (Central illustration). Complete cerebral vessel coverage was adequate in 94.6% of treated patients, and the ProtEmbo was stable for the duration of the TAVI procedure. Interference with the TAVI procedure by the ProtEmbo device was considered minimal (Table 2).
Histopathological analysis indicated that the majority of the heparin-coated mesh surface remained free of debris, and the filter pores were completely open (thrombus formation score = 0). Scanning electron microscope image analysis did not reveal any damage of the heparin coating on the mesh surface, and there was no evidence of device-related thrombogenicity or any significant embolic deposition or accumulation on the device surface. The median area of the ProtEmbo device surface containing debris was 0.121 mm2, which comprises <0.006% of the filter area (total surface area equals 2,184 mm2).
Efficacy outcome
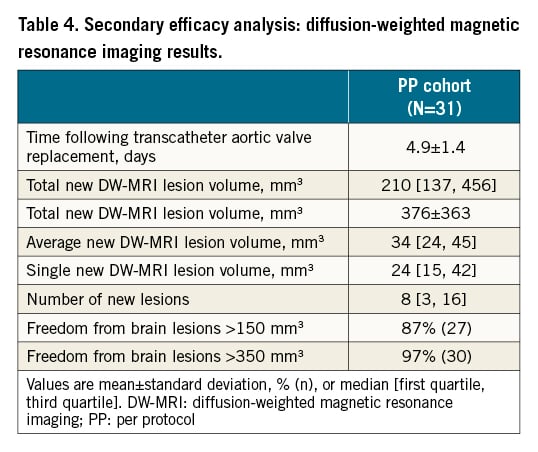
In the PP cohort there were no deaths or strokes (Central illustration). The median total new lesion volume among patients receiving treatment with the ProtEmbo device was 210 mm3 [137, 456] (Table 4). The largest single lesion volmues in each patient were all <500 mm3; the largest single lesion volume detected in any of the patients was 402 mm3; 87% of patients were free of single lesions >150 mm³; and 97% were free of single lesions >350 mm³. Supra-threshold lesion volume analysis at lesion volume thresholds of >100 mm3, >200 mm3, and >500 mm3 compared favourably with historical data from the control arm of a previous randomised controlled trial of a CEPD2.
Discussion
The PROTEMBO C Trial demonstrated that the ProtEmbo System performed as intended, meeting both primary safety and performance endpoints, and can be used safely as an adjunct to TAVI with minimal interaction. The primary safety rate was low in comparison to precedent CEPD studies and in the context of early feasibility studies, with no serious adverse events being adjudicated related to the use of the ProtEmbo. The histopathological evaluation further supported the safety and haemocompatibility of the ProtEmbo for use during TAVI.
The ProtEmbo device was easy to use with a minimal learning curve across 10 different operators performing the investigational device procedure and achieving a high technical success rate of 94.6% in this study, suggesting that the device can easily be adapted into the normal workflow of TAVI procedures. The additional contrast media and fluoroscopy time needed for use of the ProtEmbo device was negligible. The time to place the device and its stability once in place was without reported undue interference with the TAVI procedure except in 1 patient, in whom the investigational device was shifted during TAVI; however, this did not prolong the TAVI procedure. In addition, the use of the left radial artery for vascular access seems favourable in comparison with transfemoral CEPDs because interaction between the ProtEmbo and the TAVI procedure was minimal using 3 different transcatheter heart valve (THV) technologies. Adequate coverage of all 3 cerebral vessels was achieved in 94.6 % of patients treated with the ProtEmbo, which may have led to the low new lesion burden observed in patients in this study.
The efficacy results compare favourably with results from patients in the control groups of randomised controlled trials such as the REFLECT II trial of the TriGuard device2 (Keystone Heart) and the SENTINEL trial30. Although trial-to-trial comparisons should be interpreted with caution, in the REFLECT II trial, patients who did not receive embolic protection had a mean total new lesion volume of 508 mm3 compared with 376 mm3 in patients treated with the ProtEmbo. Furthermore, in our series with the ProtEmbo, no patient had a DW-MRI lesion >500 mm3 (maximum lesion size was 402 mm3), which compares favourably to the unprotected control patients in REFLECT II and SENTINEL. In the SENTINEL trial, the median total new lesion volume in all territories was 310 mm3 (interquartile range [IQR] 106-860). The median total new DW-MRI lesion volume in patients treated with the ProtEmbo was 210 mm3 (IQR 137-456). The favourable performance of the ProtEmbo compared to other CEPDs evaluated in other clinical trials should be interpreted with caution; however, these comparisons are encouraging and provide a useful starting point for the design and analysis of future clinical studies.
Limitations
The PROTEMBO C Trial was a non-randomised, single-arm study, and the results of a relatively small study cannot be directly compared to a randomised control group but do provide initial evidence for the safety and performance of the ProtEmbo device. The comparison with historical data may be affected by bias related to baseline and procedural characteristics of patients in different centres and at different times.
Conclusions
The PROTEMBO C Trial demonstrated that use of the ProtEmbo device during TAVI is safe and that it performs as intended compared to historical PG. The volume of new MRI lesions in patients treated with the ProtEmbo was low compared with historical series. A future randomised controlled trial is planned to evaluate the safety and efficacy of cerebral embolic protection when the ProtEmbo device is used during TAVI.
Impact on daily practice
Despite advancements in TAVI devices and implantation techniques, embolic stroke remains the most frequent ischaemic complication after TAVI. It is associated with significant morbidity and mortality. The ProtEmbo is a novel deflection filter device and the only CEPD that can be used through the left arm arteries. A larger randomised controlled trial is merited to further evaluate the safety and efficacy of the ProtEmbo device when used for cerebral protection during TAVI.
Acknowledgements
The authors wish to acknowledge the members of the independent data and safety monitoring board (Darren Mylotte, MD; Dana Leifer, MD; Sameer Gafoor, MD; and Isaac George, MD) for their contributions to the PROTEMBO C Trial, and Victor Jimenez, MD, for proctoring the first-time use of this investigational device.
Funding
This clinical trial was supported by an unrestricted research grant from Protembis GmbH.
Conflict of interest statement
D. Jagielak declares proctoring and lecture fees from Meril Life Sciences. M. Abdel-Wahab’s hospital receives consultancy fees and/or speaker honoraria on his behalf from Medtronic and Boston Scientific. N. Werner has received proctoring and lecture fees from Edwards Lifesciences and Medtronic. A. R. Witkowski has received proctoring and lecture fees from Edwards Lifesciences and Medtronic. M. Adam has received personal fees from Edwards Lifesciences and Boston Scientific; grants and personal fees from Medtronic; and proctoring fees from Medtronic. F. Gatto reports proctoring fees from Boston Scientific and Medtronic. T. Schmidt has received travel expenses from Protembis. The other authors have no conflicts of interest to declare.
Supplementary data
To read the full content of this article, please download the PDF.
