Abstract
Aims: We describe the first-in-human experience with a novel cerebral embolic protection device used during transcatheter aortic valve implantation (TAVI). One current challenge of TAVI is the reduction of procedural stroke. Procedural mobilisation of debris is a known source of cerebral embolisation. Mechanical protection by transient filtration of cerebral blood flow might reduce the embolic burden during TAVI. We aimed to evaluate the feasibility and safety of the Claret CE Pro™ cerebral protection device in patients undergoing TAVI.
Methods and results: Patients scheduled for TAVI were prospectively enrolled at three centres. The Claret CE Pro™ (Claret Medical, Inc. Santa Rosa, CA, USA) cerebral protection device was placed via the right radial/brachial artery prior to TAVI and was removed after the procedure. The primary endpoint was technical success rate. Secondary endpoints encompassed procedural and 30-day stroke rates, as well as device-related complications. Deployment of the Claret CE Pro™ cerebral protection device was intended for use in 40 patients, 35 devices were implanted into the aortic arch. Technical success rate with delivery of the proximal and distal filter was 60% for the first generation device and 87% for the second-generation device. Delivery times for the first-generation device were 12.4±12.1 minutes and 4.4±2.5 minutes for the second-generation device (p<0.05). The quantity of contrast used related to the Claret CE Pro System was 19.6±3.8 ml. Captured debris was documented in at least 19 of 35 implanted devices (54.3%). No procedural transient ischaemic attacks, minor strokes or major strokes occurred. Thirty-day follow-up showed one minor stroke occurring 30 days after the procedure, and two major strokes both occurring well after the patient had completed TAVI.
Conclusions: The use of the Claret CE Pro™ system is feasible and safe. Capture of debris in more than half of the patients provides evidence for the potential to reduce the procedural cerebral embolic burden utilising this dedicated filter system during TAVI.
Introduction
Transcatheter aortic valve implantation (TAVI) is emerging as a valid therapeutic option for patients with severe symptomatic aortic stenosis. Recent randomised clinical trials demonstrated TAVI to be the treatment of choice in patients with prohibitive surgical risk.1 This technique might even be regarded as an alternative, non-inferior treatment in patients at high risk for cardiac surgery.2 However, TAVI is associated with a significantly increased risk for cerebral stroke at 30 days and 12 months as compared to medical therapy or surgical valve replacement.1,2
The Placement of Aortic Transcatheter Valves (PARTNER) trial has shown that TAVI is associated with a 30-day rate of transient ischaemic attack (TIA) or stroke of 6.7% in patients at prohibitive surgical risk in comparison to a 1.7% rate in patients receiving conservative treatment, 83% of which undergoing balloon valvuloplasty.1 In patients at high surgical risk, TAVI was associated with a 30-day rate of TIA or stroke of 5.5% in comparison to 2.4% with the surgical approach.2 Besides the higher rates of apparent cerebrovascular events within the first year after TAVI, recent studies using diffusion-weighted magnetic resonance imaging have raised concerns regarding embolic events related to the procedure itself, reporting an incidence of post-procedural cerebral embolic events in up to 84% of TAVI patients.3-5 Although the clinical importance of silent cerebral embolic events has not yet been elucidated, this imaging data may reflect the higher stroke risk associated with TAVI.
Mechanical cerebral protection is frequently used in carotid interventions. Recent data suggest that this may reduce cerebral embolism and improve clinical outcomes.6-8 This would suggest that the use of mechanical, neuro-protective devices could lead to reduction of embolic burden during TAVI as well. We report on the first-in-human experience with the Claret CE Pro™ protection system (Claret Medical, Inc. Santa Rosa, CA, USA), a novel filter-based device that holds the promise of safely and effectively reducing the cerebral embolic burden in patients undergoing TAVI.
Methods
STUDY DESIGN
This study was designed to evaluate the feasibility and safety of the Claret CE Pro™ system delivering filters for embolic protection to the brachiocephalic and the left common carotid artery prior to TAVI. After written informed consent, patients were included in the study. Central and local ethics committees approved the study.
INCLUSION CRITERIA
– Patients scheduled for elective TAVI.
– Compatible left carotid artery (>3 mm) and brachiocephalic artery (>9 mm) diameters.
– Female subjects of childbearing potential with a negative pregnancy test within 48 hours prior to the study procedure
– Written informed consent.
EXCLUSION CRITERIA
– Emergency procedure.
– Carotid artery stenosis >70% in either carotid artery.
– Significant stenosis, ectasia, dissection or aneurysm at the ostium or within 3 cm of the ostium of the brachiocephalic or left carotid artery.
– Bleeding diatheses or coagulopathy or refusal of blood transfusion.
– Renal insufficiency, defined as a creatinine level >2.5 mg/dl at the time of treatment, unless subject is on chronic haemodialysis.
– Hyperthyroidism.
– Recent (within the past three months) stroke with permanent deficit or recent (within the past six months) significant gastrointestinal (GI) bleed.
– Participation in another clinical study or other medical illnesses that may cause the subject to be non-compliant with the protocol or confound the data interpretation.
– History of intolerance, allergic reaction or contraindication to any of the study medications, including heparin, aspirin, clopidogrel or to materials from which the device is constructed.
DEVICE AND PROCEDURE
The Claret CE Pro™ System (Figure 1) is designed to filter cerebral blood flow within the ostia of the brachiocephalic trunk and its right carotid branch, as well as in the left common carotid artery both originating directly from the aortic arch. The proximal filter consists of a nitinol frame designed to allow apposition within vessels measuring 9-15 mm in diameter and containing a polyurethane filter with 140 µm diameter pores. The frame is radiopaque and will expand to oppose and seal against the vessel wall when unsheathed. The proximal filter is attached to a 100 cm long catheter, and following insertion, the proprietary proximal filter is deployed in the brachiocephalic artery, followed by the delivery of a second non-proprietary filter (e.g., SpiderFX™, Covidien, Mansfield, MA, USA, or FilterWire™, Boston Scientific, Natick, MA, USA) to the left common carotid artery (Figure 2). The entire system can be delivered through a 6 Fr sheath introduced through either the brachial or radial artery of the right arm. The system is deployed immediately prior to passage of the TAVI delivery catheter through the aortic arch and into the native valve during the TAVI procedure, and is removed after removal of the TAVI delivery catheter.
All patients were pre-treated with clopidogrel 600 mg and, in case they were not on chronic acetylsalicylic acid (ASA) treatment, they received ASA 500 mg on the day before the procedure. In all patients, a standard 6 Fr sheath was placed in the right radial or brachial artery, after which heparin was given to achieve an activated clotting time above 250 seconds. Two generations of the device were used in this study, with the first generation system delivered to the aortic arch without a guidewire under direct fluoroscopic visualisation in the first seven patients. The second-generation device included the addition of a 0.014” guidewire lumen as well as a modified curve shape that included a “counter-bend” tip (Figure 1).
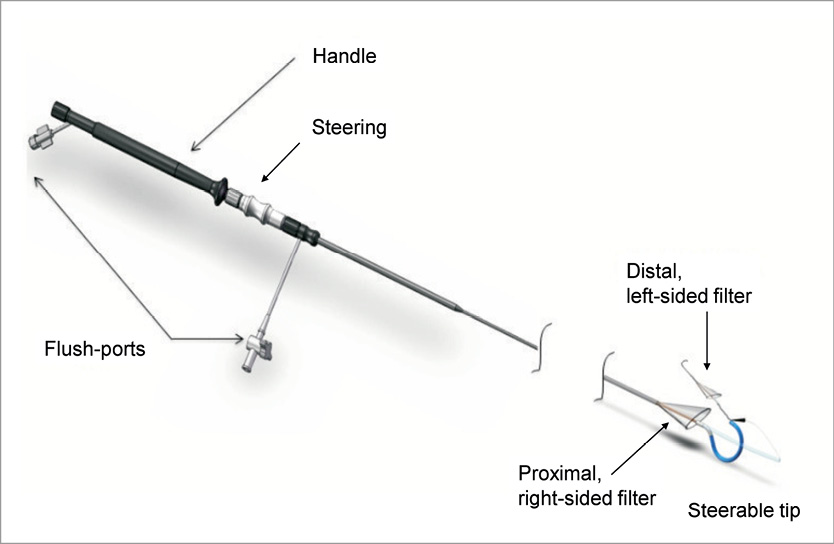
Figure 1. The Claret CE Pro™ System. The Claret CE Pro™ System consists of a proximal filter attached to a 100 cm long catheter shaft. The proximal filter is fixed within a flexible nitinol frame, which is delivered to the brachiocephalic artery within the delivery catheter. The frame is radiopaque and will expand to oppose and seal to the vessel wall when unsheathed. After delivery of the proprietary filter to the brachiocephalic artery, the system enables the delivery of a commercially available filter to the left common carotid artery. The entire system can be delivered through a 6 Fr sheath introduced from the right radial or brachial artery.
After visual inspection of the porous filter membrane, the device was flushed and retracted into the delivery catheter, and subsequently introduced into the 6 Fr sheath. With the tip of the delivery catheter positioned in the aortic arch, the proximal filter was deployed within the brachiocephalic artery under fluoroscopic guidance. The steerable, rotatable and translatable coaxial inner portion of the catheter was then extended through the proximal filter, and retrogradely advanced into the aortic arch where the distal tip was articulated in order to cannulate and place the second or distal filter in the left common carotid artery. Prior to starting the TAVI procedure, apposition of the radio-opaque proximal filter frame to the vessel wall was confirmed with angiography to ensure protection of the cerebral vascular circulation (Figure 2). TAVI was performed with the third generation Medtronic CoreValve (CoreValve Revalving Technology, Medtronic, Minneapolis, MN, USA) in 38 patients and with the Edwards SAPIEN valve prosthesis (Edwards Lifesciences, Irvine, CA, USA) in two patients.
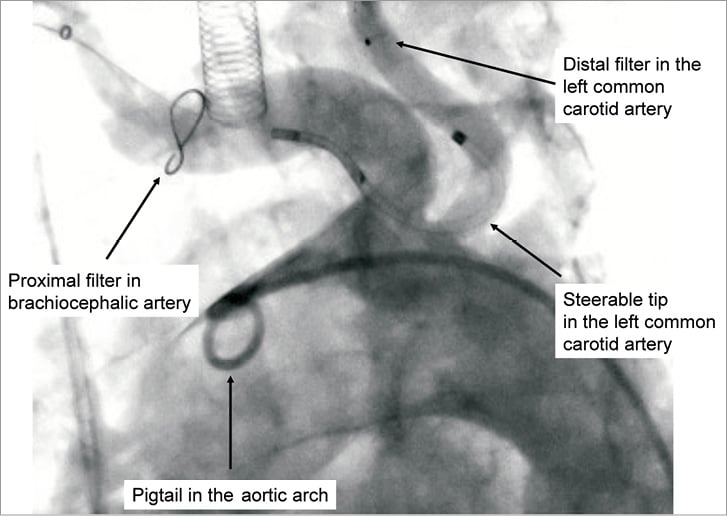
Figure 2. The Claret CE Pro™ System in situ. Representative angiography of the radiopaque protection device within the targeted vessels showing the proximal filter deployed within the brachiocephalic artery protecting the right anterior, middle and posterior circulation, and the distal filter deployed in the left common carotid artery, protecting the left anterior and middle cerebral blood supply.
ENDPOINTS AND DEFINITIONS
The primary endpoint was technical success, defined as successful delivery and retrieval of the Claret CE Pro System to the aortic arch, and, in these patients, the successful placement of the Claret CE Pro™ System filters to the brachiocephalic and left common carotid arteries followed by successful retrieval of both filters. Secondary endpoints were device-related safety outcomes including periprocedural rates of TIA, minor stroke, major stroke and device-related complications. Stroke severity was quantified according to the National Institutes of Health Stroke Scale (NIHSS) score. According to the recent consensus statement, a “rapid” post-interventional deficit was defined as a procedural event and a Modified Rankin Score ≥ 2 was defined as major stroke.9 In addition, 30-day follow-up was systematically collected for TIA, minor stroke, major stroke and device-related complications.
STATISTICS
Continuous variables are reported as mean ± standard deviation, and categorical variables are reported as n (%). Differences between first-generation and second-generation devices were based on the Student t-test for continuous variables and the chi-squared test for categorical variables. In addition, 95% confidence intervals were computed for point estimates of the primary endpoint. Statistical significance was set at the two-tailed 5% level. All analyses were conducted with IBM SPSS Statistics version 20.0.0 (IBM Corporation, Somer, NY, USA).
Results
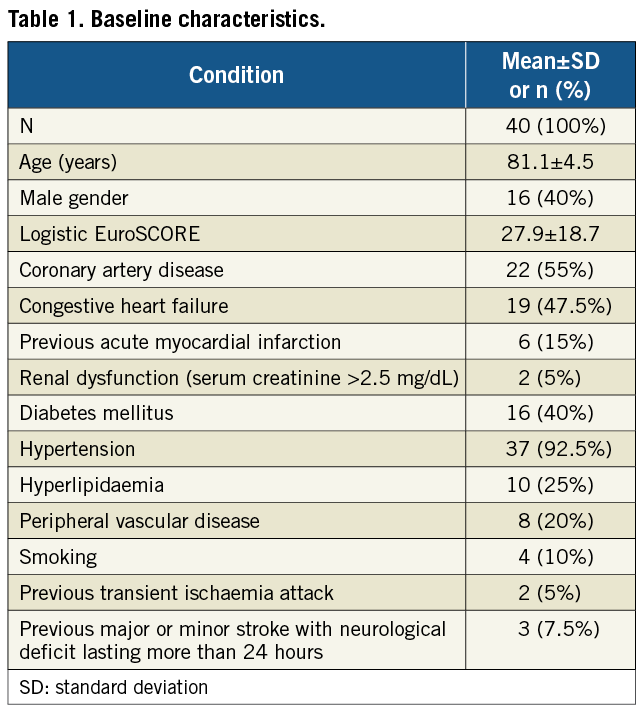
Forty subjects were enrolled between February 2010 and May 2011 at three centres in Germany and Brazil. The severity of comorbidities in this patient cohort was comparable to previous studies reflected by a mean logistic EuroSCORE of 27.9±18.7, with five patients (12.5%) having a history of cerebrovascular events. Detailed baseline characteristics are shown in Table 1.
PRIMARY, TECHNICAL OUTCOME
The access site was radial in 5 (12.5 %) and brachial in 35 (87.5 %) patients. For the protection of the left common carotid artery, a commercially available SpiderFX carotid filter was the most commonly used filter device (36 patients, 90%). Notably, there was no interference with the transfemoral valve system used in any of the device generations.
Five out of seven first-generation proximal filter devices (71%) could be delivered to the aortic arch, with one arterial spasm and one dissection of the radial artery leading to placement failures in two cases. Both filters were deployed in three of these patients, while in two patients the left carotid artery could not be accessed with the first generation device (Figure 3, Table 2).
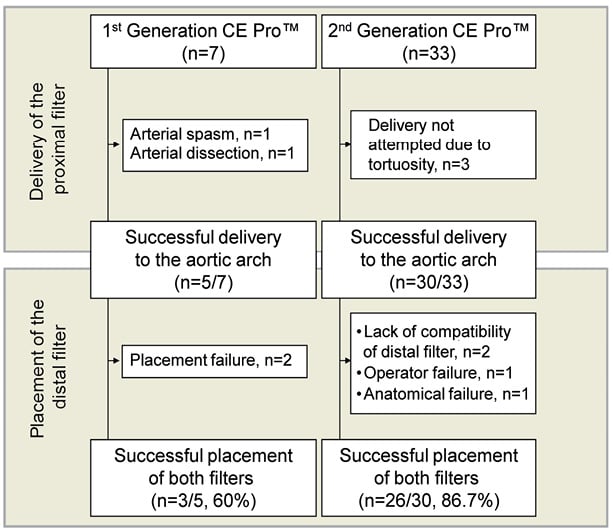
Figure 3. Technical success. Seven and 33 patients were evaluated for cerebral protection with the first- and second-generation CE Pro(TM) system, respectively. In all, 29 of the 35 systems delivered to the aortic arch were successfully placed within the brachiocephalic and left carotid artery (83%). The design of the first-generation device lacked a guidewire port and a completely steerable catheter tip, resulting in one spasm and one dissection of the radial artery. Further, in two patients the distal filter could not be placed within the left carotid artery. Therefore, technical success was low with the first-generation device (60%). The delivery of the second-generation device was not attempted in three of 33 patients, due to excessive vessel tortuosity. Two of the 30 attempted second-generation systems could not be placed bilaterally due to suboptimal compatibility of the distal SpiderFX-filter with the Claret catheter. In one case, operator failure prevented the placement of the distal filter. In another case, the left carotid artery could not be accessed. The second-generation device demonstrated, after accessing the aortic arch, a success rate of 87%.
Thirty out of the 33 second-generation filter devices were successfully delivered to the aortic arch (91%). In three cases, the brachiocephalic artery was judged to be too tortuous to make an attempt to place the device. In those cases where an attempt was made, placement of both filters was accomplished in 26 of 30 patients (87%). In two cases the distal filter could not be deployed due to incompatibility of the non-proprietary filter with the Claret catheter. In these cases pulling on the filter wire (SpiderFX 7.0 mm) led to inadvertent locking of the distal filter in the device which was prevented subsequently by slight modifications of the device design. In one case, operator failure (unintentional deformation of the device) prevented the placement of the distal filter. In only one case did the anatomy of the left carotid artery prevent the placement of the distal filter (Figure 3, Table 2).
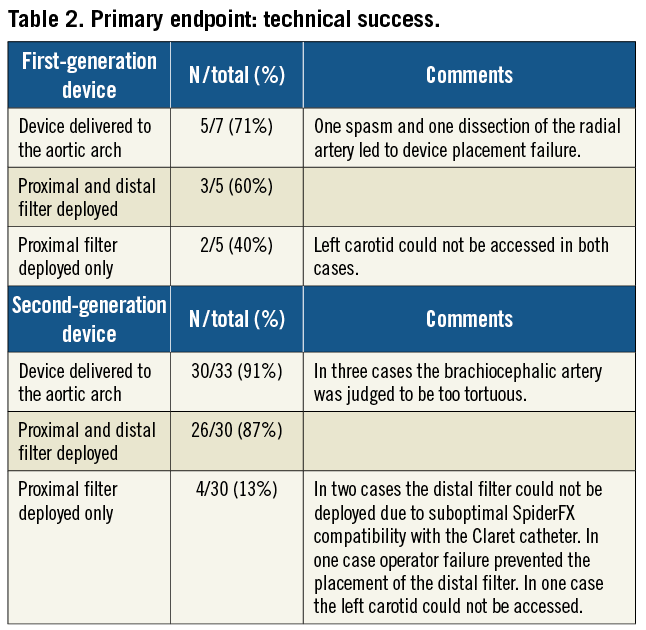
Technical success improved significantly with the use of the second-generation device (60% vs. 87%; p<0.05). As compared to the first-generation device, the second-generation system was also characterised by technical improvements resulting in significant reduction in delivery time (Figure 4). While the mean delivery time of the first-generation device was 12.4±12.1 minutes, it was 4.4±2.5 minutes for the second-generation device (p<0.05). The quantity of contrast agent used during the Claret CE Pro System deployment was 19.6±3.8 ml. Filter indwelling time varied from 25 minutes to 239 minutes, depending on the overall procedure time. Although photographic documentation of captured embolic debris was not systematically gathered, documented macroscopic evidence of captured debris was present in 19 of the 35 (54.3%) individuals where at least one filter was deployed (Figure 5).
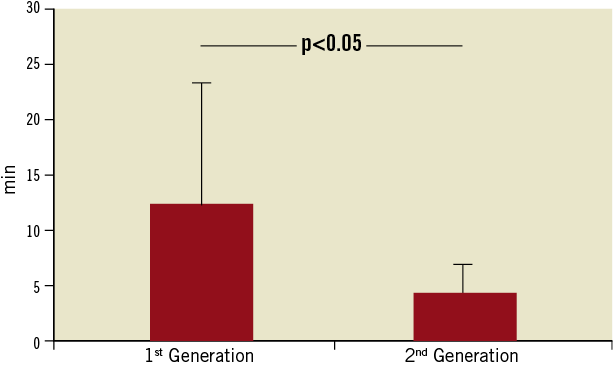
Figure 4. Delivery times of the Claret CE Pro™ System. With the development of the second-generation device, delivery to the aortic arch was guided with an 0.014” guidewire. Furthermore, control and steerability of the catheter tip was significantly improved, resulting in a significant decrease in the mean delivery time.
SECONDARY, CLINICAL OUTCOME
In terms of device-related procedural and the index hospitalisation complication rate, four events occurred. In one patient, manipulation of the (first-generation) device caused a dissection of the radial artery, which had to be treated surgically. In a second patient, a minor branch of the radial artery was ruptured, also with a first-generation device that lacked a guidewire. This led to a minor haematoma that was treated with manual compression without any clinical consequence for the patient during the follow-up period. In contrast, no device-specific procedural complications occurred with the second-generation system. However, in two cases, in relation to the procedure, a brachial pseudo-aneurysm developed after removal of the vascular sheath and following mechanical compression of the puncture site. Both were treated surgically. There were no device related complications after 30 days.
With respect to cerebrovascular events, no TIA, minor or major stroke occurred during the procedure. One patient revealed a minor stroke (NIHSS score: 2) 30 days after the procedure. Further, two major strokes (NIHSS scores: 9 and 4) were obtained four hours and 27 days after the procedure. All three patients underwent TAVI in sedation without use of general anaesthesia. The routine, immediate post-interventional neurological status in the catheterisation laboratory demonstrated no focal deficit.
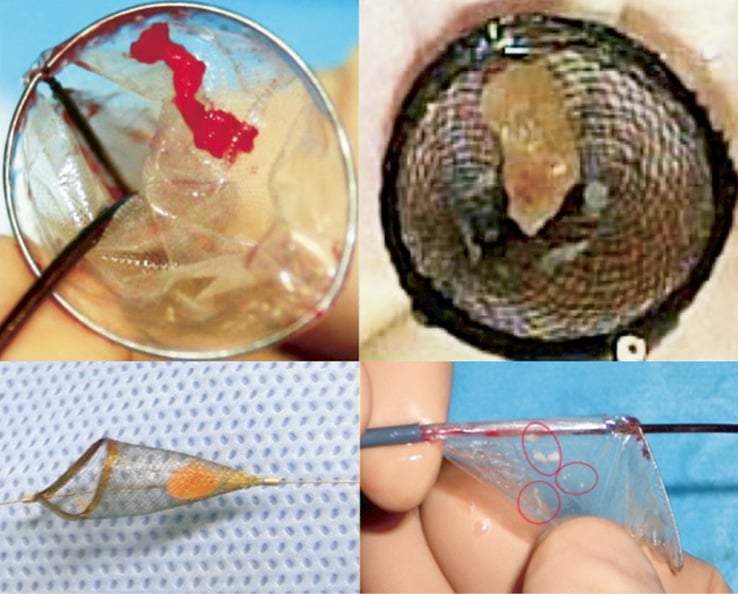
Figure 5. Representative examples of TAVI-induced liberation of debris. Representative specimens were retrieved within the proximal (right, brachiocephalic filter) and distal filters (left, carotid filter) verifying successful bilateral reduction of embolic burden with the Claret CE Pro™ system during TAVI.
Discussion
Disabling stroke is the one of the most challenging complication of the TAVI procedure and is one of the reasons to only cautiously extend the indication to lower risk cases despite the recently reported excellent outcomes in these patients.10 Stroke after TAVI, however, is not a uniform entity but may result from a variety of causes. Interestingly, only half of the neurologic events in the PARTNER trial were found being related to the procedure itself.1,2 The other half of the events occurred during the 12 months following the procedure. Recently, a “high-risk period” for cerebrovascular events was described within the first 24 hours following TAVI.11 However, post-procedural events were shown to be associated with different cardio-embolic sources. One potential source might be the bioprosthesis itself being not endothelialised and thrombogenic, and thus a potential source of cerebral embolic events.11 The other source might be the new-onset of atrial fibrillation after TAVI associated with a 3.9-fold increase of early post-procedural stroke risk in patients without history of atrial fibrillation.12
Procedural stroke, however, is most likely embolic, and is caused by mechanical shedding of debris from the aortic arch or the calcified aortic valve during device insertion, balloon angioplasty or valve deployment. In an effort to minimise the risk of procedural stroke during TAVI, embolic protection devices such as the Claret CE Pro System or the Embrella (Edwards Lifesciences, Irvine, CA, USA) deflection system have been developed.13 Data on the latter device remain limited to a four-patient pilot series.
The present study on 40 patients shows that it is safe and feasible to protect the brain bilaterally from embolic debris during endovascular repair of aortic valves by introducing a temporary filtration system (Claret CE Pro System, Claret Medical, Santa Rosa, CA, USA) into the arterial circulation via the right brachial or right radial artery.
While the first generation of the device was difficult to place, changes in the device design increased the success rate significantly. With the modified second-generation device, the proximal filters could be deployed in all cases where an attempt was made to place the device, and, in only one case the left carotid anatomy prevented the placement of the distal filter. Nevertheless, future modifications of the device will have be developed to prevent potential compatibility related failure such as the locking of the distal filter in the device by pulling on the filter wire before deployment.
Specific device related adverse events in the first generation, such as perforation and artery dissection, could be prevented in the second generation by guiding the device with a 0.014” guidewire. Radial access should be preferred over brachial access to avoid local complications such as the observed pseudo-aneurysms and to minimise the potential risk for puncture related ischaemia.
Given the low complication rate of the second generation bilateral filter protection system, the current data suggest that the use of such a system could be highly beneficial for patients undergoing endovascular aortic repair. Such a system may even be used in surgical aortic valve repair with reported procedural stroke rates of 2-3%.14
Notably, a filter system placed during the procedure will not be effective in prevention of post-procedural early and late embolism, which will have to be addressed by other means.
The main question is, how many patients should be protected and how many events may be prevented by applying a bilateral brain protection system during endovascular repair of aortic valves?
There are two published manuscripts on procedural overt stroke in TAVI patients. They report on 354 individuals with a stroke rate of 0.6-10%.5,15 Otherwise, there are preliminary reports showing procedural stroke rates between 2.5 and 5.0%.16-18
On the one hand, mechanical protection may not always result in 100% protection. Bonati et al report on a substantial rate of silent cerebral embolic events in patients undergoing “protected” carotid intervention.19 And, with the Embrella deflection device, Nietlispatch et al report on a cerebral embolic event in one of four patients in “protected” balloon aortic valvuloplasty.13 Thus, despite this, though we demonstrate as a proof-of-concept the retrieval of embolic debris in more than half of our patients, further studies will have to investigate the exact degree of protection provided by the use of the Claret CE Pro™ system.
Still, on the other hand, looking only at overt stroke may strongly underestimate the need for protection of the brain during aortic valve procedures. Despite the fact that the diagnosis of overt stroke has been clearly defined,9 it is known that the number of hits3-5 significantly exceeds the number of overt strokes and, the clinical relevance of these hits is not yet fully understood. In clinical practice, patients may experience a change in personality or of their cogitation without manifest neurological symptoms that can be related to subclinical procedural embolism after such procedures.20 These questions will have to be addressed in the future, and, may then unravel an even higher clinical and ethical need for protection during TAVI, during aortic valvuloplasty, and, during surgical aortic repair. Thus, further evidence is eagerly awaited to clarify the role of this technology in routine clinical practice: e.g., a randomised trial will have to look at the reduction of immediate overt procedural stroke, but other data from controlled trials and carefully conducted registries are needed concerning the reduction, as well as the clinical importance, of silent cerebral ischaemia during TAVI. In addition, these will have to investigate subclinical psychological and personality changes that may occur later, after the procedure. Cost-effectiveness data, as well, would also allow expanding the use of this technology to high-risk patients undergoing TAVI.
Conclusion
The current study shows that the use of the Claret CE Pro system to protect the brain from embolic debris during endovascular repair of aortic valves is feasible and safe. The capture of debris was successfully documented in at least half of the patients, and represents a proof-of-concept for this approach in protecting patients from stroke during these procedures. The potential benefit of this method may be even higher, since the prognostic damage caused by subclinical, procedural, cerebral hits needs to be further elucidated in specifically designed trials.
Acknowledgements
We wish to thank Dr. Giuseppe Biondi-Zoccai of the University of Modena and Reggio Emilia, Modena, Italy, for his editorial support.
Conflict of interest statement
The authors have no conflict of interest to declare.
