Abstract
Aims: The aim was to assess the safety and efficacy of percutaneous patent foramen ovale (PFO) closure with the Premere (St. Jude Medical, Inc., St. Paul, MN, USA) device.
Methods and results: This is a prospective clinical and echocardiographic follow-up of 264 patients who underwent transcatheter PFO closure with planned implantation of the Premere device. Implantation was successful in 263 patients (99.6%). Complete closure demonstrated echocardiographically occurred in 92.7%. The 30-day adverse event rate was 5.4% (atrial fibrillation in six, pericardial effusion in three, acute coronary syndrome in two, pseudoaneurysm/fistula formation at the access site in two patients and device dislocation in one patient). At a mean follow-up of 19.3 months (±14.2 months) stroke or transient ischaemic attack (TIA) occurred in nine (3.5%) and thrombus formation on the left atrial anchor in one (0.4%) patient.
Conclusions: These data demonstrate that PFO closure with the Premere closure device is safe and effective. Complication rates and efficacy are similar to previously studied devices.
Introduction
Transcatheter PFO closure has been performed routinely over the past two decades1-4. Multiple closure devices have been used with good efficacy and safety. However, few devices are specifically designed for PFO and most devices do not allow device adjustments during implantation to better suit the PFO tunnel length or highly variable thickness of the interatrial septum. To this effect, the Premere PFO closure device (St. Jude Medical, Inc., St. Paul, MN, USA) was designed. It allows adjustments during the procedure depending on the tunnel length and has a very low profile on the left atrial side, thereby potentially minimising the risk of device-related thrombi. Its use was first described in the first-in-man efficacy and safety (CLOSEUP) trial in 2003. In this trial, a high procedural success, efficacy and low complication rate was demonstrated5. Though it is routinely used, only limited data have been published since. Donti et al showed successful implantation in 15 patients without any complications within the first postprocedural month6. Rigatelli et al demonstrated improvement of migraine symptoms after closure with the Premere device7. Reyes et al reported no device-related complications after implantation in 11 patients8. A mid-term follow-up study including 70 patients with no complications was recently published by Rigatelli et al9. Kleber et al showed successful implantation in 72 patients with a low peri- and postprocedural adverse event rate10. Thaman et al compared three different PFO closure devices. After implantation of the Premere device in 38 cases, residual shunting could be observed in 18.5%11. Bissessor et al reported no recurrent strokes/TIA during a median follow-up of 11 months12. These studies demonstrate that, though the Premere device has been used in some countries and has achieved the European conformity (CE) mark, data regarding its safety and efficacy are limited. We therefore present the follow-up results of 263 patients after implantation of a Premere closure device for PFO closure at a single centre. The aim was the safety and efficacy assessment of this device for PFO closure.
Materials and methods
Patient population
The study is a prospective registry including clinical and echocardiographic follow-up. Data of the implantation procedures and follow-up was analysed and a standardised questionnaire sent to all patients annually. Collected patient data were recorded using Microsoft Excel (Microsoft Corp., Redmond, WA, USA). Between February 2003 and February 2010, 1,710 patients underwent transcatheter PFO closure at our institution. Overall, 347 patients received a Premere device (20%). Fifty patients were excluded from our study due to prior publication within the CLOSEUP trial as were 34 patients who received a Premere device due to residual shunting after previous implantation of a different PFO closure device. In 264 patients an attempt at Premere device implantation was made, and in 263 cases a Premere device was successfully implanted as the first PFO closure device. Indications for PFO closure were cryptogenic stroke or transient ischaemic attack, peripheral arterial embolism thought to be related to paradoxical embolism, history of decompression sickness and migraine. The choice of the implanted device was based on device availability and operator preference. No device-specific inclusion or exclusion criteria were used. The presence of a right-to-left shunt through a PFO was ascertained by contrast transoesophageal echocardiography (TEE) or contrast transthoracic echocardiography (TTE). All patients had given written informed consent prior to the procedure. The study was approved by the Institutional Review Board.
Device characteristics
The Premere PFO closure device (Figure 1) consists of two cross-shaped nitinol flexible anchors. The right-sided anchor is positioned between two membranes of knitted polyester. The left-sided anchor is not covered to reduce the surface of the device and thereby potentially facilitate endothelialisation and minimise the risk of thrombus formation. A flexible polyester-braided tether connects the two nitinol anchors and allows the left and right anchor arms to conform independently to variations in septal thickness and tunnel length without any distortion of the atrial anatomy. One end of the tether is linked to the left-sided anchor, the other to the external side of the delivery system. The right-sided anchor is connected to the delivery guidewire. The Premere closure device is currently available in two sizes: 20 and 25 mm. Both require an 11 Fr delivery sheath (St. Jude Medical, Inc., MN, USA).
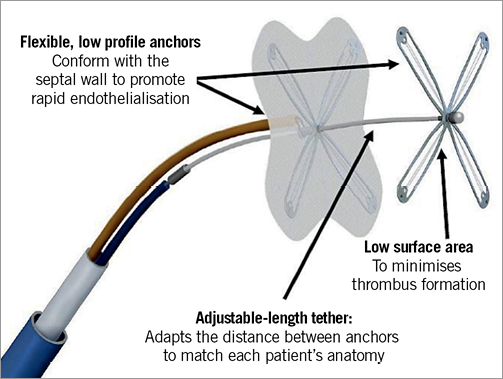
Figure 1. The Premere PFO closure device.
The device can be retrieved during the procedure at any time prior to its release. In case of misplacement after device release, retrieval with the Premere retrieval basket (St. Jude Medical, Inc., St. Paul, MN, USA) is possible.
PFO closure
All patients received intravenous antibiotic prophylaxis before and after device implantation (in the absence of a penicillin allergy, 1.5 g IV ceftriaxone pre- and post-procedure). The procedure was performed under local anaesthesia in all but one case. Intravenous propofol or midazolam were used for sedation. Procedures were generally performed under transoesophageal echocardiography (TEE) and fluoroscopy guidance. Though intraoperative TEE and balloon sizing is not essential for Premere device implantation, we perform both procedures routinely for PFO closures to facilitate device selection and positioning. After establishing femoral venous access, a bolus of 10,000 units of heparin was administered to all patients. A 5 Fr multipurpose catheter (Cordis Corporation, Johnson & Johnson, Warren, NJ, USA) was advanced over a J-tipped 0.035 inch guidewire (Cook Medical, Bloomington, IN, USA) into the right atrium and then inserted through the PFO under fluoroscopic guidance. The 0.035 inch guidewire was then placed into the left upper pulmonary vein. Subsequently, balloon sizing (NMT Medical, Inc., Boston, MA, USA) was performed to determine the optimal device size. An 11 Fr delivery sheath (St. Jude Medical, Inc., St. Paul, MN, USA) was positioned in the left atrium and used to advance the Premere PFO closure system.
Under transoesophageal and fluoroscopy guidance, the left-sided anchor was released and attached to the interatrial septum by pulling the polyester tether connecting both anchors. After retracting the introducer sheath into the right atrium, the right atrial anchor was deployed. To optimise conformation to the individual anatomy and minimise tissue distortion, the distance between the two anchors was adapted by pulling the tether under echocardiographic control.
Satisfactory device position was confirmed by TEE and/or fluoroscopy imaging, the right atrial anchor subsequently locked and detached releasing the device and the delivery sheath removed.
Endocarditis prophylaxis was recommended according to ACC/AHA guidelines for six months after device implantation. From the beginning of the study to February 2007 the standard antiplatelet therapy consisted of 100 mg of aspirin and 75 mg of clopidogrel daily for six months. From February 2007 onwards, aspirin 100 mg po daily was used for six months and clopidogrel 75 mg daily for three months.
Follow-up
All patients were instructed for follow-up either at our centre in Frankfurt or with the referring cardiologist, the choice being that of the patient. Follow-up examinations were recommended at 1, 3, 6 and 12 months and yearly thereafter.
Patients were recommended to undergo contrast supported transoesophageal echocardiography (TEE) and ECG combined with a cardiac history and physical examination at one and six month follow-up to evaluate the device position, presence of a residual shunting and the existence of thrombi as well as potential complications. A transthoracic echocardiogram (TTE), ECG cardiac history and physical examination were recommended at three-month follow-up. Finally, a 12 month follow-up was recommended for patients who were found to have a residual shunt on previous examinations. A standardised questionnaire was used to interview the patients annually. Primary endpoints were incidences of the following: 1) stroke recurrence; 2) TIA recurrence; 3) haemopericardium; 4) new onset atrial fibrillation; 5) device-related thrombus formation; 6) death by any cause.
Statistical analysis
Data analysis regarding procedural complications (30-day adverse event rate) was performed on an intention-to-treat principle (all patients were included in whom the implantation of a Premere device had been attempted, whether a Premere had been successfully implanted or a different device was eventually used). Analysis regarding late complications and efficacy was performed including only patients who received the Premere device.
Statistical analysis was performed with GraphPad InStat for Windows (version 3.10; GraphPad Software, Inc., La Jolla, CA, USA). Data are expressed as means with standard deviation and range. Categorical variables were compared with the chi-squared test.
Results
Patient population
Baseline characteristics are shown in Table 1. In all but three patients, the closure indication was a previously documented thromboembolic event. In one patient, the PFO closure was performed as primary prophylaxis for thromboembolic events anticipated with diving. One patient suffered from severe migraine and a third patient was diagnosed with a very large PFO with left-to-right shunting while hospitalised because of acute dyspnoea and thoracic pain. Atrial dilation caused by atrial fibrillation had resulted in severe left-to-right shunting through the PFO and in heart failure. Figure 2 demonstrates the number of thromboembolic events for each patient before and after PFO closure.
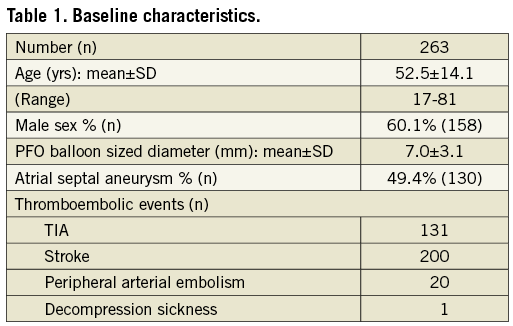
Figure 2. Number of thromboembolic events per patient before and after PFO closure.
Procedural details
Device implantation was successful in 263 (99.6%) of attempted interventions (264 patients). In one case, the Premere device prolapsed into the right atrium before release. Retrieval was uneventful and a different PFO closure device was implanted successfully. With the exception of one procedure during which the TEE probe was not tolerated despite moderate sedation and therefore general anaesthesia was required, all were conducted under local anaesthesia. TEE was performed in all but three patients (1.1%), who did not tolerate the TEE probe. In these patients correct device position was assessed using fluoroscopy and TTE only. The mean stretched PFO diameter measured by balloon sizing was 7.0±3.1 mm (range 1.6-24.8 mm). Tunnel length was measured by TEE according to the American Society of Echocardiography and the Society of Cardiovascular Anesthesiologists (ASE/SCA) guidelines13. A detailed overview of PFO anatomy was published by Saremi et al in 200814. The mean tunnel length was 10.0±3.7 mm (range 2.2-24.7 mm).The 20 mm device was implanted in 176 (66.9%) cases, whereas the 25 mm device was used in 87 (33.1%) patients. The mean PFO diameters in the 20 mm and 25 mm device groups were 5.6±2.1 mm (range 1.6-12.7 mm) and 9.9±2.9 mm (range 3.1-24.8 mm), respectively. The mean procedural time was 33.9±13.7 min (range 10-120 min) and the mean fluoroscopy time 4.8±4.0 min (range 1.2-33.3 min).
30-day procedure and device-related event rate, including immediate procedural events
The 30-day adverse event rate was 5.4% (14/260 patients). Three (1.1%) patients experienced an intraprocedural adverse event. An acute coronary syndrome occurred in two cases. In the first patient, dyspnoea and angina pectoris occurred immediately after the procedure. A non-ST-segment elevation myocardial infarction was diagnosed and the patient recovered without further events during the hospital stay. Severe coronary heart disease was subsequently diagnosed and the patient referred to cardiac surgery. Coronary air embolism caused transient ST-elevation in the other case. Coronary angiography was performed immediately and showed no pathological findings. The patient recovered after treatment with tirofiban and aspirin without any sequelae. In one case, a 20 mm Premere device dislocated after device release. Percutaneous retrieval of the device was uneventful and a 25 mm Premere device was implanted.
New onset atrial fibrillation was reported in six patients (2.3%) after a mean post-procedural time of 0.5±0.2 months (range 0.2-0.8 months). In two cases, it spontaneously converted to sinus rhythm. Pulmonary vein isolation was performed in one patient. Electrical cardioversion was performed in one patient in whom long-term anticoagulation was initiated due to atrial fibrillation recurrence. One patient was treated with rate control and long-term anticoagulation, while one patient was treated with rate control and aspirin/clopidogrel only.
Pericardial effusions were seen in three cases (1.2%) in one of which, due to haemodynamic significance, pericardiocentesis was required. In the remainder, spontaneous resolution was documented. One patient developed a pseudoaneurysm at the access site 14 days after device implantation. This resolved spontaneously. Surgical intervention was performed in one case on an arteriovenous fistula at the access site three days after the procedure. The operation was uneventful and recovery without sequelae. In both cases inadvertent arterial puncture was assumed.
No strokes or transient ischaemic attacks occurred within 30 days of device implantation.
Long-term follow-up
Follow-up data ≥1 month was available for 260 (98.9%) patients. Three (1.1%) patients were lost to follow-up due to relocation. The mean follow-up duration was 19.3±14.2 months (range 0-55 months).
Strokes
Strokes occurred in six (2.3%) patients; two major (National Institute of Health stroke score [NIHSS] of >3) and four minor (NIHSS ≤3). The annual stroke rate after PFO closure was 1.4% (six recurrent strokes/5,009 patient-months). The average duration from the procedure to the occurrence of a stroke was 7.4±4.4 months (range 1.4-13.5 months). All patients were examined by a neurologist. Four of the strokes were confirmed by CT or MRI scans. One patient experienced a neurological deficit lasting >24 hours without confirmation by neuroimaging and one patient suffered from acute monocular vision loss with ophthalmoscopically confirmed retinal ischaemia. Complete resolution could not be observed during follow-up. In five of the stroke patients, complete closure at the time of recurrent stroke was confirmed. Thrombus formation was excluded by TEE following the stroke in three cases, in one case TEE showed no thrombus formation 14 days prior to the event and in another case, two months prior to the event. One patient did not attend further TEE follow-up examinations after one month follow-up. A residual shunt had been observed at that time. A definitive stroke mechanism could not be identified in either of the patients. Five patients were treated with antiplatelet agents (three patients received aspirin, one patient clopidogrel and one patient aspirin and clopidogrel) and, due to possible paradoxical embolism, the patient with stroke and incomplete closure was treated with long-term anticoagulation.
Transient ischaemic attacks
A TIA was observed in three (1.2%) cases during follow-up. In one case the TIA occurred after stopping antiplatelet treatment with aspirin three years after PFO closure. Complete PFO closure was confirmed at the time of the incident and aspirin prescribed again. In the second case, the TIA occurred two months post-procedure. Complete closure had been documented at the one-month TEE follow-up. No further medication apart from aspirin and clopidogrel were prescribed. In the third case a TIA combined with thrombus formation on the left atrial anchor was noticed 21 months post-procedure. Complete PFO closure had been documented at one-month follow-up. The TIA prompted the performance of a TEE at an outside institution and a thrombus was reportedly seen on the left atrial anchor. Anticoagulation with phenprocoumon was initiated immediately. However, due to an increase in thrombus size despite anticoagulation, the closure device was removed and the PFO closed surgically. The operation was uneventful. TEE seven months after surgery showed no residual shunting or thrombus. Importantly, this patient had also experienced atrial fibrillation 20 days after closure.
Device-associated thrombus
Thrombus formation on the left atrial anchor was seen by TEE in one case (0.4%), as described above. The annual event rate for thrombus formation was 0.2% (one patient with thrombus formation/5,009 patient-months).
Implantation of further devices
In six (2.3%) patients the implantation of a second PFO device was necessary during follow-up. In three of these cases severe residual shunting was the reason for re-intervention and in one patient multiple defects of the atrial septum. In addition, in one case, device embolisation discovered 47 days after implant required re-intervention and implantation of a second device. In this case catheter-based device extraction was uneventful and an AMPLATZER® Septal Occluder (AGA Medical Corporation, Plymouth, MN, USA) was implanted successfully but with residual shunting. In one patient the left-sided anchor of the Premere device was found to be positioned partially in the right atrium, which resulted in residual shunting. In five cases a second Premere device was implanted, in four of which further follow-up demonstrated complete PFO closure. Therefore, of the six patients who required implantation of further devices, residual shunting remained in two cases.
Other events
One patient suffered from atrial fibrillation and cardiac decompensation due to tricuspid valve insufficiency one year after PFO closure. He underwent tricuspid annuloplasty. The intraoperative findings showed incomplete PFO closure. The closure device was replaced by a cardiac patch during this operation. Another patient suffered from new onset atrial fibrillation one year post-procedure. This resolved spontaneously. The patient with PFO closure due to severe migraine reported complete resolution of migraine headaches after PFO closure at one-month follow-up.
Death
Overall, five (1.9%) patients died during follow-up. Two patients died of cancer, one patient died during coronary bypass operation and in two cases the cause of death is unknown.
Closure rates
During a mean follow-up time of 19.3±14.2 months (range 0-55 months) complete PFO closure was achieved in 241/260 (92.7%) patients. Closure rates for the individual stretched diameters are listed in Figure 3. Complete closure could be observed in 96.2% of patients with a median diameter smaller than 6.7 mm. A significantly lower closure rate of 89.2% was seen in PFO with a median diameter of equal to or greater than 6.7 mm (p<0.03). Complete closure was achieved in 93.2% (164/176) in the 20 mm device group and in 91.7% (77/84) in the 25 mm device group. This difference was not statistically significant. In patients with an atrial septal aneurysm in addition to the PFO, complete closure occurred in 91.5% (118/129) compared to 93.9 % (123/131) in those without an atrial septal aneurysm. Once again, this difference was not statistically significant.
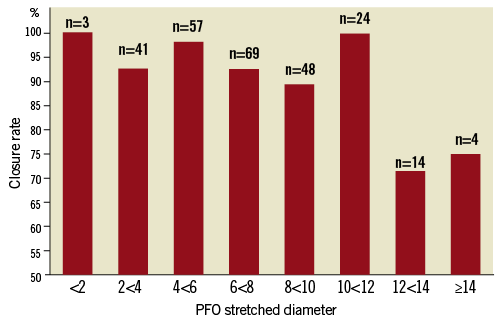
Figure 3. Closure rates by PFO stretched diameter.
Discussion
In this report we present the acute and long-term results of transcatheter PFO closure with the Premere closure device in 263 patients. The focus was the investigation of procedural success, incidence of adverse events (including atrial fibrillation and thrombus formation) and efficacy (complete closure rates and recurrent thromboembolic events).
The immediate procedural and 30-day adverse event rate (including the occurrence of atrial fibrillation) is comparable to previously reported event rates experienced with the implantation of other devices. Khairy et al, in a systematic review of 1,355 PFO closures with various devices, reported a procedural major complication rate of 1.5%. This included death, haemorrhage requiring transfusion, need for surgical intervention and cardiac tamponade. Minor complications, including potentially device-related arrhythmic events occurred in 7.9%15. Similarly, in a single centre study of 525 patients who underwent fluoroscopy-guided PFO closure with eight different devices (predominantly the AMPLATZER PFO occluder) the immediate procedural event rate was 2.5%. This included device embolisation, symptomatic air embolisation, vascular access complications and pericardial tamponade, none of which resulted in long-term sequelae16.
New onset atrial fibrillation is a common complication after percutaneous PFO closure. Staubach et al evaluated the incidence of new onset atrial fibrillation after PFO closure in 1,349 patients. Atrial fibrillation occurred in 3.9% after a mean follow-up of 38 months17. Likewise, after a median follow-up of 20 months, the incidence of atrial fibrillation was 7% in a study by Spies et al18. This is similar to the incidence of atrial fibrillation (3.1% at a mean follow-up of 19.3 months) in our study. It is unclear whether patients with thromboembolic events and presence of a PFO are more likely to develop atrial fibrillation than a healthy population. However, it is conceivable that the mere presence of a PFO increases the likelihood of atrial fibrillation. Alternatively, some patients whose thromboembolic events were attributed to paradoxical embolism may in fact have experienced these events as a result of undetected paroxysmal atrial fibrillation with a PFO as an innocent bystander. To this effect, a recently published study by Bonvini et al compared the occurrence of atrial fibrillation after percutaneous PFO closure with a population of medically-treated patients with PFO19. The incidence of atrial fibrillation was similar in both groups. The only significant risk factor for new onset atrial fibrillation was the presence of a large PFO. A relationship between device type and new onset atrial fibrillation was reported by Taaffe et al in a randomised trial which compared three PFO closure devices (AMPLATZER®, Helex® [W. L. Gore & Associates, Inc., Flagstaff, AZ, USA] and CardioSEAL®-STARflex® [NMT Medical, Inc., Boston, MA, USA]). A notable difference between the individual devices was seen. New onset atrial fibrillation occurred in 5%, 1.4% and 0.9% of the patients treated with the CardioSEAL-STARflex device, AMPLATZER device and Helex device, respectively20. A recent publication suggests that PFO closure may be associated with a reduction of new onset atrial fibrillation. Jarral et al showed an anti-arrhythmic effect in a meta-analysis of six publications including 2,570 patients21. It can be hoped that further prospective randomised trials will clarify whether atrial fibrillation seen after PFO closure is related to the device or PFO itself, or whether it is entirely unrelated to either.
The stroke rate in our study is similar to that reported in other studies. The incidence of recurrent thromboembolic events after PFO closure varies depending on device type, duration of follow-up and study design. A recently published review by Staubach et al systematically analysed 21 publications regarding long-term complications22. Recurrent embolic events such as stroke or TIA were reported in up to 5.8% of the patients during follow-up of as long as 45.6 months. In this review, newer devices were associated with fewer device-related complications. In the systematic review by Khairy et al the incidence of recurrent thromboembolic neurologic events at one year varied between 0 and 4.9% depending on the study15.
Thrombus formation is a rare but serious complication. We observed thrombus formation associated with the PFO device in one case (0.4%) during a mean follow-up of 19.3 months. In an attempt to minimise the likelihood of thrombus formation, the Premere device has a particularly small left-sided surface area. Krumsdorf et al analysed the incidence of thrombus formation after transcatheter closure of interatrial shunts in a large patient population23. It was found in 2.5% after PFO closure in 593 patients during a mean follow-up time of 36 months for all interatrial closure devices (PFO+atrial septal defect [ASD] closure devices). This publication comprises patient data collected from 1992 to 2003 with the use of early closure devices. The incidence of thrombus formation in these bulkier devices may be higher compared to newer devices with lower profiles. Staubach et al compared the incidence of thrombus formation in studies with early and new PFO closure devices22. Thrombus formation was seen in up to 7% of early devices. Newer devices showed no cases of thrombus formation. This study was limited by a small patient population and limited follow-up time with newer devices compared to devices that had already been used over a longer period. Finally, Wahl et al reported thrombus formation in 0.8% of 525 patients after PFO closure. All thrombi eventually resolved after anticoagulation16.
Migraine with aura has been reported to be associated with the presence of PFO24. In our study one PFO closure was performed due to severe migraine. This patient showed complete resolution of migraine one month after PFO closure. The current literature indicates that PFO closure might reduce the intensity and frequency of migraine headaches25. However, conclusions regarding the severity or frequency of migraine attacks cannot be made from this study.
Closure rates in our patient population compare well with those reported in the current literature (between 86% and 100% depending on the device type and follow-up time)22. Although higher closure rates are desirable, the Premere device is frequently selectively used for patients with long PFO tunnels due to the unique adjustable distance between the left- and right-sided anchors. With more rigid fixed distance devices such as the AMPLATZER PFO occluder, tissue distortion is inevitable and residual shunting, particularly in patients with long tunnels or thick interatrial septum, may be more common if these devices were studied specifically under these anatomic conditions. Rigatelli et al demonstrated successful implantation of the Premere closure device in 70 patients with a closure rate of 98.5%, including 20 patients with hypertrophic or lipomatous rims9. Further studies with specific focus on anatomic PFO features may elucidate the advantages and disadvantages of various devices regarding the incidence of residual shunting. In our study closure rates were lower in patients with stretched PFO diameters >6.7 mm. Likewise, Shafi et al investigated predictors for residual shunting after PFO closure26. A predisposition for residual shunting was found in patients with large PFO sizes. Due to the somewhat lower closure rates in large PFO, at our institution closure is no longer performed with the Premere device for PFO with large stretched diameters.
Limitations
First, not all patients underwent follow-up, thus the incidence of adverse events may be under-represented. More importantly, all potential sources of error inherent to a retrospective non-randomised non-blinded study apply, including, among others, sampling bias. Importantly, the selection of any specific device including the Premere device for PFO closure was not guided by any prespecified criteria. Further, the Premere device was not compared to any other devices in a controlled or randomised fashion. Therefore, neither can definitive statements regarding the merits of this device compared to others be made, nor can a conclusion be made regarding device suitability for various anatomical PFO configurations.
Conclusion
Our data demonstrate that PFO closure using the Premere closure device is safe and effective. Closure rates are similar to those reported with other devices. Major complications are rare. The incidence of device-associated thrombus formation is low and the incidence of new onset atrial fibrillation similar to other devices. Most importantly, recurrent thromboembolic events are no more, and perhaps less, likely than those reported with other devices.
Conflict of interest statement
All authors have no conflict of interest to declare.
