
Abstract
Aims: A second iteration of a sirolimus-eluting stent (SES) that has a biodegradable PLGA polymer coating with an electrografting base layer on a thin-strut (80 µm) cobalt-chromium platform (BuMA Supreme; SINOMED, Tianjin, China) has been developed. This first-in-man trial aimed to assess the efficacy and safety of the novel device.
Methods and results: This randomised, multicentre, single-blinded, non-inferiority trial compared the BuMA Supreme SES versus a contemporary durable polymer zotarolimus-eluting stent (ZES) in terms of angiographic in-stent late lumen loss (LLL) at nine-month follow-up as the primary endpoint. A total of 170 patients were randomly allocated to treatment with either SES (n=83) or ZES (n=87). At nine-month angiographic follow-up, in-stent LLL was 0.29±0.33 mm in the SES group and 0.14±0.37 mm in the ZES group (pnon-inferiority=0.45). The in-stent percent diameter stenosis and the binary restenosis rate of the two treatment arms were similar (19.2±12.0% vs. 16.1±12.6%, p=0.09, and 3.3% vs. 4.4%, p=1.00, respectively). At 12-month clinical follow-up, there was no difference between treatment arms with regard to the device-oriented composite clinical endpoint (4.9% vs. 5.7%; p=0.72).
Conclusions: The PIONEER trial did not meet its primary endpoint in terms of in-stent LLL at nine-month follow-up. However, this result did not translate into any increase in restenosis rate or impairment in 12-month clinical outcomes.
Abbreviations
BP: biodegradable polymer
CD: cardiac death
CI-TLR: clinically indicated TLR
DES: drug-eluting stent
DoCE: device-oriented composite endpoint
DS: diameter stenosis
LLL: late lumen loss
OCT: optical coherence tomography
QCA: quantitative coronary angiography
SES: sirolimus-eluting stent
TIMI: Thrombolysis In Myocardial Infarction
TLR: target lesion revascularisation
TV-MI: target vessel myocardial infarction
ZES: zotarolimus-eluting stent
Introduction
Second-generation drug-eluting stents (DES) have been developed with more biocompatible durable polymer coatings on thin-strut stents and have demonstrated improved safety relative to first-generation DES1-3. Biodegradable polymer (BP)-DES were developed to reduce long-term polymer-related adverse effects4. Several studies have shown improvement of clinical outcomes with BP-DES as compared to durable polymer DES4.
The BuMA™ Supreme (SINOMED, Tianjin, China) sirolimus-eluting stent (SES) consists of a thin-strut cobalt-chromium platform (80 µm) with a thin (200 nm) electrografted base layer, to which a biodegradable top coat is firmly adhered. The base layer is bonded to the stent surface and both anchors and aligns the molecule of the biodegradable top layer. This prevents the bioactive coating from cracking and delamination upon delivery and expansion of the stent (Figure 1). The BuMA Supreme SES has a pharmacokinetic polymer degradation/drug release profile where the top coat releases the drug relatively rapidly such that sirolimus release from the stent surface is nearly complete by 28 days after implantation. Less than 1 ng/mg of the drug is maintained in the arterial wall at 60 days after implantation (Figure 2). Several studies in humans and animal models have demonstrated the efficacy and safety of its predecessor, the BuMA stent, which used a stainless steel platform of 100 µm5.
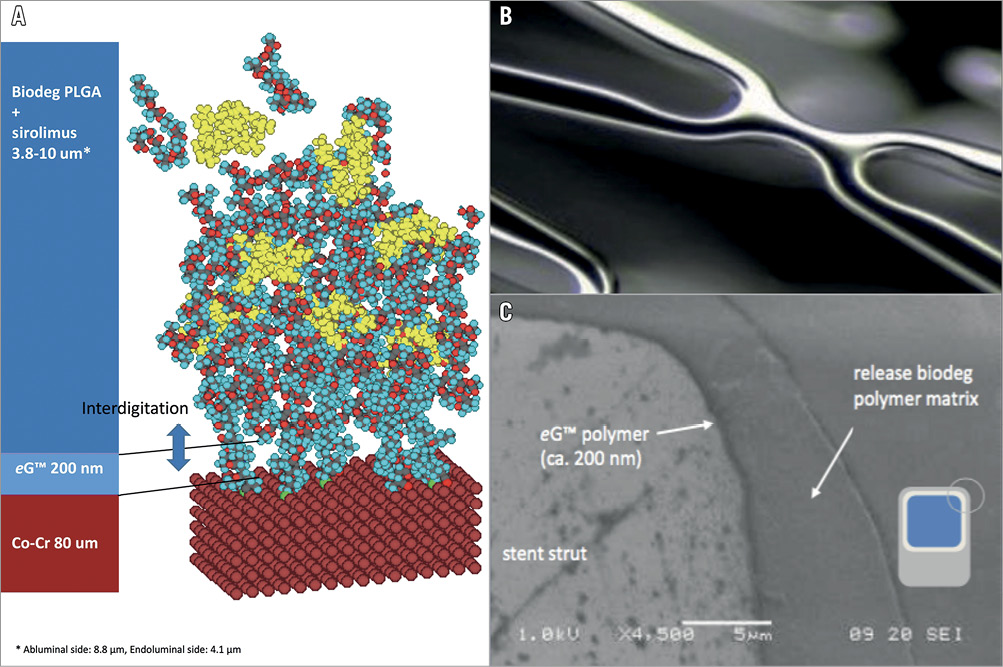
Figure 1. The BuMA Supreme stent design. A) Electrografting coating technology (eG): polymer chains (200 nm) of poly n-butyl methacrylate covalently bound by electrografting to an electronically polished thin-strut (80 µm) cobalt-chromium stent platform. B) Link between cells. C) Electron microscopy (×4,500) showing strut of BuMA Supreme (left), electrografted base layer (eG) (mid) and polymer matrix of PLGA containing sirolimus.
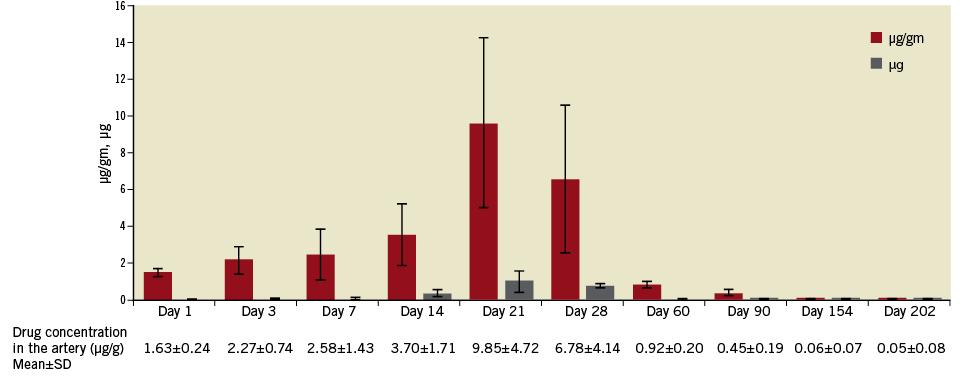
Figure 2. Pharmacokinetics of the artery after implantation of the BuMA Supreme SES (porcine model). Stented artery segment sirolimus concentration peaked up to 21 days. Peak correlates with polymer degradation and with blood levels.
The present first-in-man study assessed nine-month angiographic and 12-month clinical endpoints to evaluate the efficacy and safety of the BuMA Supreme SES versus a durable BioLinx™ (Medtronic, Santa Rosa, CA, USA) tripolymer-coated (Resolute-type) zotarolimus-eluting stent (ZES). The latter device has shown favourable outcomes in randomised trials6,7.
Methods
STUDY DESIGN AND POPULATION
The PIONEER trial is a multicentre, single-blinded, two-arm 1:1 randomised trial, which was designed to demonstrate non-inferiority of the BuMA Supreme SES versus a Resolute-type ZES in terms of nine-month angiographic in-stent late lumen loss (LLL). In addition, secondary clinical endpoints were assessed at 12-month follow-up.
Patients were eligible for study enrolment if they met eligibility criteria, the details of which are listed in the Supplementary Appendix. Briefly, patients who presented with stable or unstable angina or silent ischaemia with one or two separate, de novo target lesions in a reference vessel of 2.5-4.5 mm were enrolled in the current trial. Patients with evolving myocardial infarction, bifurcated target lesion, target lesion in left main artery, aorto-ostial target lesion and restenotic target lesion were not eligible for the current trial.
The PIONEER study complied with the Declaration of Helsinki, medical ethics committees at each participating institution approved the trial, and all patients provided written informed consent.
STUDY DEVICES AND IMPLANTATION PROCEDURE
The BuMA Supreme SES is a DES with a rapid exchange balloon-expandable catheter delivery system. The device consists of an extremely thin (200 nm) electrografted (eG) base layer of poly(n-butyl methacrylate) (PBMA), perpendicularly and covalently bound to an electronically polished thin-strut (80 µm) cobalt-chromium stent platform. The base layer forms a thin brush that interdigitates with a 3.8-10 μm-thin top layer –a blend of a biodegradable polylactide-co-glycolic acid (PLGA) polymer and the drug sirolimus (drug concentration: 120 µg/cm² of stent surface). The PLGA polymer layer is designed to resorb in six weeks (Figure 1)8.
Resolute-type ZES (Resolute Integrity® or Onyx™; both Medtronic, Minneapolis, MN, USA) were used in the control group. Resolute Integrity is made from a round cobalt-chromium wire (91 µm) and Resolute Onyx from a swaged-shape core wire (81 μm) that consists of a platinum-iridium core surrounded by cobalt-chromium. Both iterations of Resolute-type ZES elute zotarolimus over a six-month period6,7,9-11. Interventional procedure was performed and dual antiplatelet therapy was administered according to current clinical guidelines.
ENDPOINTS
The primary study endpoint was in-stent LLL at nine months after stent implantation, as assessed by off-line quantitative coronary angiography (QCA) in at least two paired matched views. Secondary angiographic endpoints included acute lumen gain, in-segment LLL at nine months, minimum lumen diameter post procedure and at nine months, percent diameter stenosis (DS) post procedure and at nine months, and binary restenosis (DS ≥50%) at nine months. All measurements were performed in-stent, in-segment, and on the 5 mm proximal and distal stent margins.
Secondary clinical endpoints included: 1) acute device success, 2) procedural success, and 3) a device-oriented composite endpoint (DoCE), defined as the composite of cardiac death, target vessel myocardial infarction (TV-MI), and clinically indicated target lesion revascularisation (CI-TLR). Each definition is described in the Supplementary Appendix. Stent thrombosis was defined according to Academic Research Consortium (ARC) definitions12 and was assessed as a secondary endpoint. MI was defined according to the criteria of the third universal definition13 except for MI type 4a (periprocedural MI), for which the SCAI definition14 was used.
An independent clinical events committee adjudicated the clinical endpoints; an independent data safety monitoring board supervised the trial. Angiographic follow-up was performed at nine months. Patients were clinically followed at 1, 9, and 12 months. The trial was monitored by an independent clinical research organisation (Cardialysis, Rotterdam, the Netherlands).
QUANTITATIVE CORONARY ANGIOGRAPHY ANALYSIS
Off-line QCA analysis was performed by an independent core laboratory (Cardialysis, Rotterdam, the Netherlands) using the CAAS system (Pie Medical Imaging, Maastricht, the Netherlands) according to standard protocols. If the target lesion was revascularised at any time between baseline and nine months, the pre-revascularisation angiogram was analysed.
STATISTICAL ANALYSIS
Categorical variables were reported as counts and percentages, and between-group differences were assessed by χ2 test or Fisher’s exact test, as appropriate. Continuous variables were presented as mean±SD and compared with a t-test. Unless otherwise specified, a two-sided p-value <0.05 was considered statistically significant. The statistical analysis was performed by SAS, version 9.3 (SAS Institute, Cary, NC, USA). Sample size calculation of the trial is described in the Supplementary Appendix.
Results
BASELINE AND PROCEDURAL CHARACTERISTICS
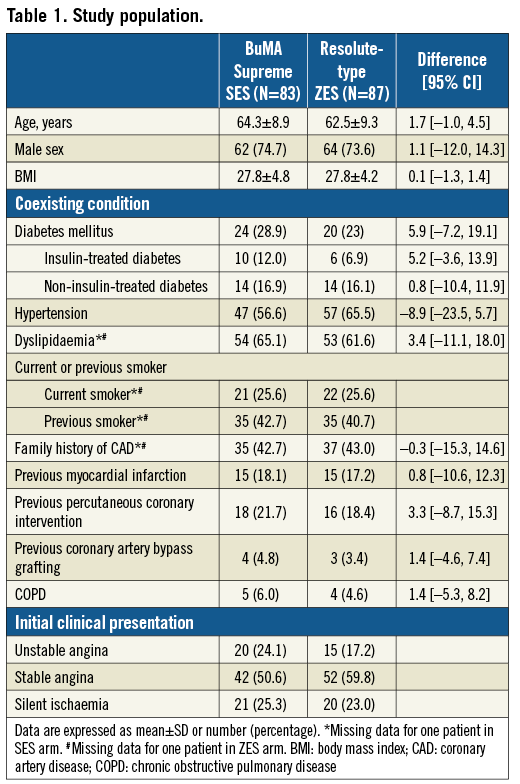
From April 2015 to January 2016, 170 patients were enrolled and randomly assigned to treatment with the BuMA Supreme SES (83 patients, 95 lesions) or Resolute-type ZES (87 patients, 101 lesions) (Figure 3). Patient demographics and clinical characteristics are shown in Table 1. Details of the interventional procedures are presented in Supplementary Table 1.
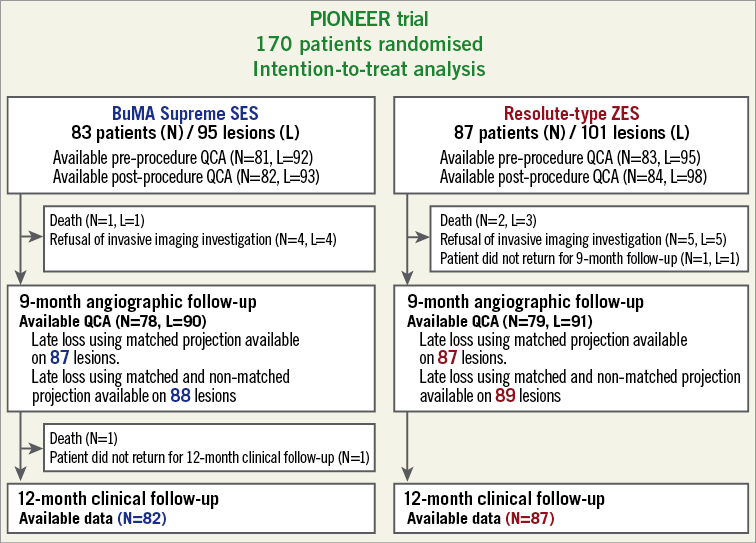
Figure 3. Study flow chart.
DEVICE AND PROCEDURAL SUCCESS
Device success was achieved in 96.8% (90/93) and 94.9% (94/99) of lesions in the BuMA Supreme SES group and the Resolute-type ZES group, respectively. Seven lesions (two in the BuMA Supreme SES group and five in the Resolute-type ZES group) were treated with a non-study stent. One lesion in the BuMA Supreme SES group had a post-procedural residual stenosis ≥30% (Supplementary Table 1).
NINE-MONTH ANGIOGRAPHIC FOLLOW-UP
In the BuMA Supreme SES and the Resolute-type ZES groups, analyses of follow-up angiography were available in 94.7% (90/95) and 90.1% (91/101) of lesions, respectively (Figure 3). The primary endpoint of in-stent LLL was 0.29±0.33 mm in the BuMA Supreme SES group versus 0.14±0.37 mm in the Resolute-type ZES group (pnon-inferiority=0.45) (Table 2, Figure 4), which means that non-inferiority was not met. Nevertheless, this did not translate into a significant difference in the rates of in-stent or in-segment binary restenosis (Figure 5).
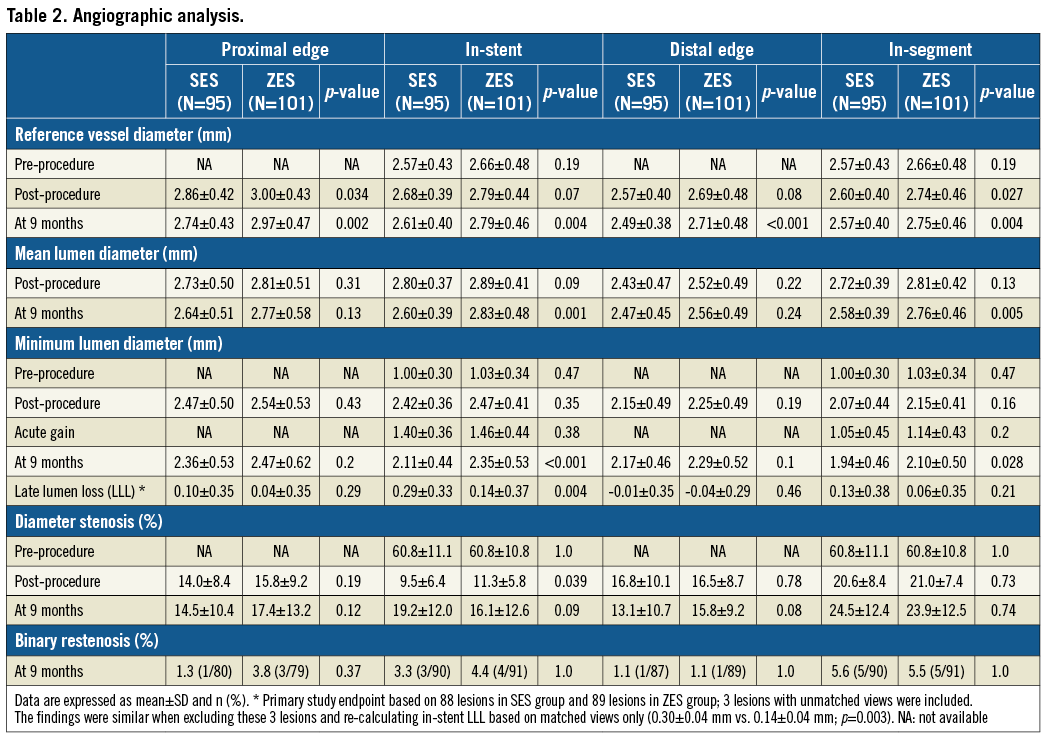
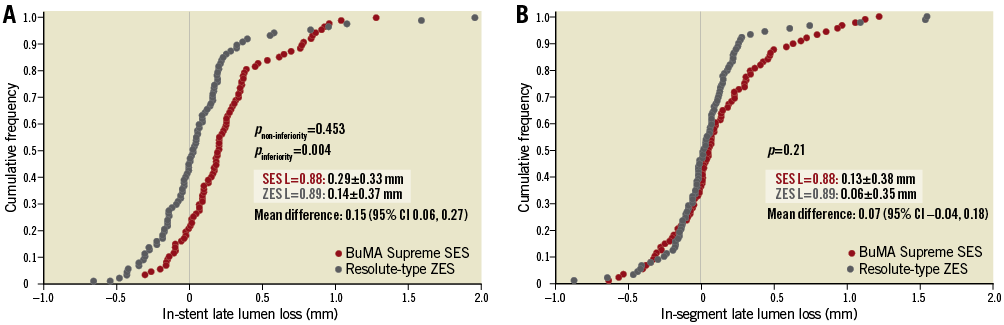
Figure 4. Cumulative frequency distribution curves of in-stent late lumen loss and in-segment late lumen loss at nine-month angiographic follow-up. A) In-stent late lumen loss. B) In-segment late lumen loss. Three LLL measurements based on unmatched views were included in this analysis (BuMA Supreme SES: 88 lesions, Resolute-type ZES: 89 lesions).
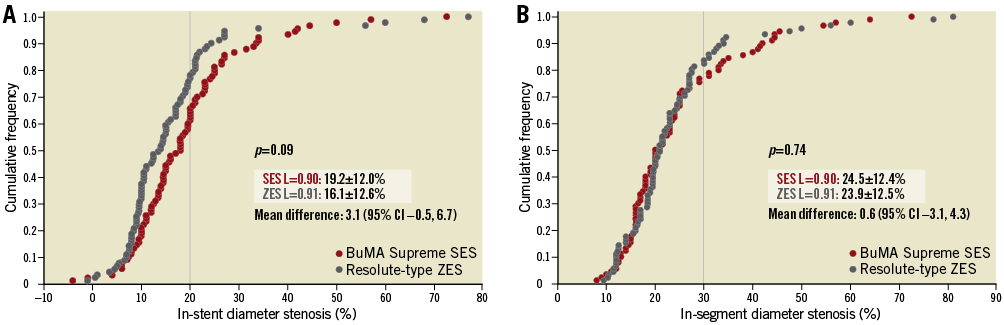
Figure 5. Cumulative frequency distribution curves of in-stent percent diameter stenosis and in-segment percent diameter stenosis at nine-month angiographic follow-up. A) In-stent percent diameter stenosis. B) In-segment percent diameter stenosis.
TWELVE-MONTH CLINICAL OUTCOMES
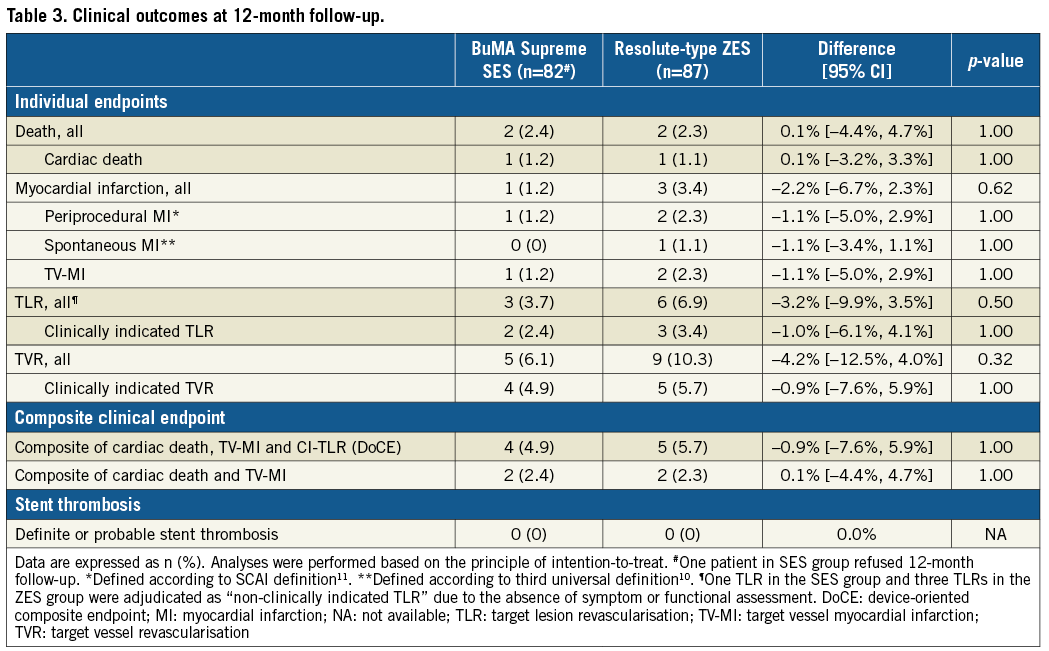
The 12-month clinical follow-up was available for 82 (98.8%) patients of the SES group and 87 (100%) patients in the Resolute-type ZES group (Figure 3). The incidence of DoCE was 4.9% (4/82) in the BuMA Supreme SES group and 5.7% (5/87) in the Resolute-type ZES group (p=1.00). CI-TLR was required in 2.4% (2/82) of the BuMA Supreme SES group versus 3.4% (3/87) of the Resolute-type ZES group (p=1.00). In both stent arms, there were no cases of definite or probable stent thrombosis (Table 3).
Discussion
The main findings of the PIONEER trial are the following: 1) the criterion for non-inferiority of the primary angiographic endpoint (in-stent LLL) was not met; 2) nevertheless, in-stent and in-segment %DS as well as the binary restenosis rate did not differ significantly between stent groups; and 3) the rates of the DoCE were similar in both treatment arms.
LATE LUMEN LOSS
In the present trial, the LLL of the BuMA Supreme SES (0.29±0.33 mm) was numerically larger than anticipated based on the previous study of its predecessor (0.24±0.33 mm)15. Reasons for this discrepancy could reside in differences in study background (e.g., patients, lesion types, lesion length, procedural characteristics such as the degree of acute gain and residual stenosis) and in dissimilar methodologies of the angiographic core laboratories. Moreover, the LLL in the Resolute-type ZES group of the present trial (0.14±0.37 mm) was lower than the LLL observed in the Resolute first-in-man (FIM) trial (0.22±0.27 mm) which, in addition, was analysed by another core lab using a different methodology16. It might be related to differences in baseline procedure. Acute gain (in-stent) of Resolute ZES in the current trial was lower than the one in the Resolute FIM trial (1.46±0.64 mm vs. 1.93±0.45 mm), whereas residual percent stenosis in the Resolute ZES group in the current trial was greater than the one in the Resolute FIM trial (11.3±5.8% vs. 3.36±8.54%). Mauri et al reported that decreased acute gain and increased residual stenosis were associated with increased LLL (estimated effect [mm]: 0.17 with acute gain [per mm], and –0.0097 with residual percent stenosis [per 1%])17. The differences in these parameters might be related to the difference in LLL between the two trials.
When PIONEER-like lesions, treated with a Resolute-type ZES, were selected from the Resolute all-comers trial, the angiographic LLL at 13 months, analysed by the core lab of the present study, was 0.19±0.26 mm7. Considering this, the relatively low LLL for the second and third iterations of the Resolute ZES may be seen in a different light and are less surprising.
OTHER ANGIOGRAPHIC PARAMETERS AND CLINICAL EFFICACY
Although in the current trial the BuMA Supreme SES did not meet the non-inferiority criterion in in-stent LLL, there was no between-group difference in in-segment percent DS (25±13% vs. 24±13%). For both DES groups the in-segment percent DS was within the range of data seen with DES that have recently been approved by the Food and Drug Administration (FDA), as has been reported by the ESC/EAPCI task force on the evaluation of coronary stents in Europe in their executive summary for the European Union18. In addition, the 12-month DoCE rates were comparably low for both treatment arms (4.9% vs. 5.7%), and in both treatment arms there was no case of definite or probable stent thrombosis.
THE CLINICAL SIGNIFICANCE OF LLL AND REGULATORY PERSPECTIVE
In the early days of balloon angioplasty, there was no quantitative method to assess the respective contribution of constrictive remodelling and neointimal proliferation. With the advent of quantitative angiography, late lumen loss, defined as a change in minimum lumen diameter between post-procedure and follow-up, was developed as a continuous variable to define restenosis and has for more than a quarter of a century remained the gold standard for regulatory bodies in the assessment of the “antirestenotic” efficacy of coronary devices19.
In the bare metal stent era, the LLL in absolute value was paradoxically doubled (0.65±0.57 mm) as compared to balloon angioplasty (0.32±0.47 mm), although minimal lumen diameter, binary restenosis rate, and target lesion revascularisation rate were improved20. LLL of a metallic coronary stent reflected exclusively the neointimal proliferation as the stent prevented constrictive vascular remodelling21. After the emergence of DES, LLL was reduced to a relatively low value, but the “near eradication” of intra-stent neointima was not per se a criterion of device safety. In the RAVEL trial, the first-generation CYPHER® (Cordis, Cardinal Health, Milpitas, CA, USA) SES had no LLL at all (-0.01±0.33 mm), but the increase in late and very late adverse events with this type of DES showed clearly that LLL was a rather unsuitable parameter to predict the efficacy and long-term safety of this device22,23. A very low LLL may indeed reflect a delayed and incomplete healing process with uncovered and malapposed struts, only seen on optical coherence tomography (OCT)24. Despite these facts, LLL remained a standard measure of the performance of new coronary stents and scaffolds18.
Angiographic LLL has been used for evaluating the process of neointimal hyperplasia and late constrictive remodelling in clinical trials with balloon angioplasty and stents, because the LLL was considered a robust continuous parameter that required a smaller sample size than the traditional binary restenosis. However, in the current era of DES, the clinical significance of comparing DES with very low LLL is debatable. Pocock et al, in data from a pooled analysis, showed that, within a range of relatively low LLL values (up to 1.0 mm), differences in LLL were not associated with a significant difference in the target lesion revascularisation rate18,25. In contrast, a mild or moderately (certainly not an excessively) increased LLL might be favourable in terms of completeness of stent coverage24.
The BuMA Supreme stent was designed to elute 92% of the drug in 28 days and leave a drug level in the artery wall 60 days after implantation of less than 1 ng/mg –a concentration that is below the therapeutic level of prevention of neointimal progression, allowing early neointimal coverage (Figure 2). In the BuMA-OCT randomised trial at three-month follow-up, the BuMA stent had a more favourable neointimal coverage than the PLA polymer-based EXCEL SES (JW Medical Systems, Weihai, China), which is characterised by drug elution during a much longer period of time (180 days)8. The Endeavor® zotarolimus-eluting stent (E-ZES) (Medtronic) was similarly designed to complete drug elution within two weeks26. Interestingly, although E-ZES had a greater in-stent LLL compared to the TAXUS™ paclitaxel-eluting stent (PES) (Boston Scientific, Marlborough, MA, USA) in the ENDEAVOR IV randomised control trial (0.67±0.49 mm vs. 0.42±0.50 mm, p<0.001), E-ZES was non-inferior to PES with rates of target vessel failure 6.6% versus 7.1%, respectively (pnon-inferiority<0.001)27. It is notable that regulatory bodies in many countries, such as the FDA in the USA, approved the E-ZES holding clinical outcomes in great account.
The ultimate treatment goal of stenting a narrowed coronary segment is to re-establish optimal hyperaemic blood supply to the subtended myocardium, which today can be quantified by a functional test such as fractional flow reserve (FFR). Future trials and regulatory bodies may embody a combined angiographic and functional approach as criteria for approval. Thus, there is currently a need to establish an optimal parameter to validate stent performance that should precisely reflect improvements in functionality and reductions in adverse clinical event risk.
Limitations
The study was powered for the primary endpoint only; clinical outcomes are only hypothesis-generating. The sample size calculation was based on data of the first-in-man study of the Resolute ZES, which was analysed by a different core laboratory.
Conclusions
The trial did not meet its primary endpoint of non-inferiority in in-stent LLL at nine-month angiographic follow-up. Nevertheless, several other angiographic parameters such as percent diameter stenosis and binary restenosis rate were similar for both stents. One-year clinical event rates, although obtained in a relatively small patient population, were comparably low with both devices. Both this and future trials will further explore whether or not the unique pharmacokinetic properties of this stent impact on long-term outcomes.
| Impact on daily practice Nine months after the implantation of the novel BuMA Supreme sirolimus-eluting stent, the trial did not meet its primary endpoint of non-inferiority in terms of angiographic late lumen loss. Nevertheless, several other angiographic parameters and clinical endpoints were similar to the comparator device, suggesting that, in the current drug-eluting stent era, the clinical significance of the parameter late lumen loss may be limited and that its suitability for predicting the efficacy and safety of novel devices is debatable. |
Acknowledgements
The authors would like to acknowledge the invaluable assistance of the members of the data safety monitoring board (DSMB), H. Boersma (Chairman), Erasmus University Medical Center, Rotterdam, H. Suryapranata, Radboud University Nijmegen Medical Center, and J. Ottervanger, Isala Klinieken, Zwolle, and the members of the clinical events committee (CEC), E. McFadden, Cork University Hospital, Scot Garg, Royal Blackburn Hospital, and Benno J.W.M. Rensing, St. Antonius Hospital, Nieuwegein.
Funding
This study was funded by SINOMED, Beijing, China.
Guest Editor
This paper was guest edited by Adnan Kastrati, MD; Deputy Director of Department Cardiovascular Diseases, Deutsches Herzzentrum München, Munich, Germany.
Conflict of interest statement
C. von Birgelen has been an unpaid consultant to several device manufacturing companies; Thoraxcentrum Twente has received institutional research funding from AstraZeneca, Biotronik, Boston Scientific, and Medtronic. P.W. Serruys, M. Sabaté, and Y. Onuma are members of the Advisory Board for Abbott Vascular. P.W. Serruys is a scientific medical consultant. The other authors have no conflicts of interest to declare. The Guest Editor has no conflicts of interest to declare.
Supplementary data
Supplementary Appendix. Details of the protocol and the procedural results in the PIONEER trial.
Supplementary Table 1. Interventional procedure.
To read the full content of this article, please download the PDF.
