Abstract
Aims: The INSPIRON-I trial is a first-in-man evaluation of the safety and efficacy of the Inspiron drug-eluting stent, a sirolimus-eluting stent with abluminal biodegradable polymer coating and thin cobalt-chromium alloy.
Methods and results: This is a randomised, multicentre comparison between Inspiron and a stent with the same metallic structure but without polymer coating or drug elution (Cronus). The primary objective was to evaluate the in-segment late loss (LLL) at six months. Secondary endpoints included percent in-stent obstruction as measured by intravascular ultrasound (IVUS) at six months and major adverse cardiac events (MACE). Fifty-eight patients were enrolled (60 lesions), 39 for Inspiron and 19 for Cronus. Baseline clinical and angiographic characteristics of both groups were similar. At six months, the in-segment LLL was reduced in the Inspiron group compared to the control group (0.19±0.16 mm vs. 0.58±0.4 mm, respectively; p<0.001), as well as the percent neointimal obstruction (7.8±7.1% vs. 26.5±11.4%; p<0.001). At two-year follow-up, incidence of MACE was similar between groups (7.9 vs. 21.1%, respectively; p=0.20), with lower target lesion revascularisation for Inspiron (0 vs. 21.1%, respectively; p=0.01) and no stent thrombosis.
Conclusions: Sirolimus eluted from an abluminal biodegradable polymer on a cobalt-chromium alloy proved effective in reducing restenosis at six months.
Introduction
Earlier-generation drug-eluting stents (DES) were highly effective1 in reducing in-stent neointimal formation and therefore in minimising the occurrence of restenosis. However, they appear to induce a small but sizeable thrombogenic response, leading to late and very late stent thrombosis2. Permanent polymers have been implicated in suboptimal polymer biocompatibility, persistent inflammatory reaction, and delayed healing observed after DES3,4. In addition, thick-strut stents, such as first-generation DES, have been demonstrated to be more thrombogenic than identical thin-strut comparators in ex vivo and experimental models5. Newer DES stent technologies have been developed to resolve these issues, such as the use of stents with thinner struts and the use of biodegradable coatings to improve biocompatibility. These properties might confer an additional benefit in terms of re-endothelialisation and restenosis6,7, particularly if the coating is applied only at the abluminal surface of the stent, leaving the luminal side of the struts free of drug and polymeric coating.
The Inspiron® (Scitech Medical, São Paulo, Brazil) sirolimus-eluting stent (SES) uses an ultra-thin L-605 cobalt-chromium alloy with a 75 μm strut thickness platform coated with a matrix of a biodegradable polymer and sirolimus applied to the abluminal surface, leading to a significantly lower concentration of drug in the vessel. We report herein the first-in-man prospective, randomised (2:1), single-blinded, multicentre trial that compared the novel Inspiron stent against an otherwise identical bare metal stent (BMS).
Methods
STUDY DESIGN AND POPULATION
Patients were enrolled at four centres in Brazil, from January 2009 to December 2010. Patients were randomised in a 2:1 basis for percutaneous coronary intervention (PCI) using the novel Inspiron DES or a BMS. Balloon angioplasty and stent implantation were performed according to standard technique.
Patients were eligible for the study if they were aged 18 years or above, had symptoms of angina or ischaemia (inducible or spontaneous). Angiographic inclusion criteria included non-occlusive de novo coronary lesions <20 mm in length and reference diameter of 2.5 to 3.5 mm. Exclusion criteria were cardiogenic shock, left ventricular ejection fraction <30%, lesions requiring two stents, ostial lesion location, major surgery within six weeks before planned stenting procedure, stenosis ≥50% of the left main coronary artery, angiographic evidence of thrombus or poor distal flow at the lesion site, lesion at a junction of a side branch with a diameter ≥2 mm, staged procedure planned within three months of the index procedure, known allergy to the study medications including sirolimus, and pregnancy (present, suspected, or planned). The study complied with the Declaration of Helsinki and was approved by all institutional ethics committees. All patients provided written, informed consent for participation in the trial. The INSPIRON-I trial was registered at the United States National Institute of Health at www.clinicaltrials.gov: NCT01093391.
STUDY STENTS
The Cronus® BMS and Inspiron® SES (both by Scitech Medical) use an ultra-thin L-605 cobalt-chromium alloy, with 75 μm strut thickness. The stent was designed in corrugated rings, with a maximum end angle between the rods of 45 degrees in its expanded form. These rings are made of short cells (1.1 mm), which allows for increasing the number of cells and which increases the homogeneity of metal cover in angulated and curved segments. The binding between rings is performed using short-shaped waveform links (65 μm) which provides a circumferential maximum diameter area of approximately 6 mm, in order to allow access to lateral branches. In the Inspiron® DES this platform is combined into a novel formulation of a SES with an abluminal biodegradable polymer. This includes a blend of poly(L-lactic acid) (PLLA) and poly(DL-lactic-co-glycolic acid) (PDLLGA) which is degraded by hydrolysis and enzymatic action to monomeric acids, and eliminated from the body through the tricarboxylic acid cycle that releases 80% of the drug within 30 days (data from Scitech Medical, São Paulo, Brazil) (Figure 1).
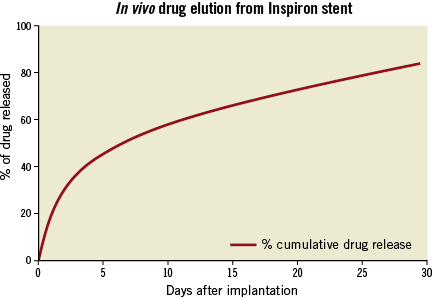
Figure 1. Drug release kinetics of the Inspiron sirolimus-eluting stent.
CLINICAL FOLLOW-UP
Baseline and procedural characteristics were recorded in a dedicated database for all patients. Adverse events were assessed in the hospital, and clinical follow-up was performed at 1, 6, 9, 12 and 24 months. Additional clinical follow-up is planned at yearly intervals up to five years. Angiographic and intravascular ultrasound (IVUS) follow-up were performed at six months.
STUDY ENDPOINTS AND DEFINITIONS
The primary endpoint of the study was in-segment late lumen loss at six months by quantitative coronary angiography (QCA). Secondary endpoints included in-stent late loss, binary restenosis rate, percentage diameter stenosis, and in-stent percentage volume obstruction on IVUS. Secondary clinical endpoints were a composite of major adverse cardiac events (MACE), including cardiac death, Q-wave or non-Q-wave myocardial infarction, clinically driven surgical or percutaneous revascularisation of the target lesion or vessel at 30 days, six months, 12 months, and 24 months after the index procedure. All of the serious adverse events were submitted to an independent adjudication committee. QCA and IVUS analyses were conducted by an independent core laboratory (Cardiovascular Research Center Angiographic and IVUS Core Laboratory, São Paulo, Brazil).
STATISTICAL ANALYSES
The sample size was estimated based on the primary endpoint of in-segment late lumen loss at six months and was calculated to detect a minimum difference of at least 0.25 mm late luminal loss between the stents tested assuming a common standard deviation of 0.35 mm with a significance level (alpha) of 0.05 and statistical power (beta) of 80%6,7. Considering the rate of loss for control angiography of 20% of the initial population, we calculated that the final total sample number for this project was 60, divided into two groups: SES (n=40) and BMS (n=20).
All data were analysed on the per-treatment evaluable population, and according to the received treatment. Discrete variables are reported as frequencies (%), and continuous variables as mean±SD. Student’s t-tests were used for continuous variables and chi-squared or Fisher’s exact tests for categorical variables. For all analyses, a two-sided p-value 0.05 was considered statistically significant. All analyses were conducted using the statistical package R version 2.13.0 (R Core Team, Vienna, Austria) and SPSS 20 (IBM Corp, Armonk, NY, USA).
Results
BASELINE CHARACTERISTICS
A total of 59 patients were randomised. One patient did not receive the study stent because the target vessel was shown to be totally occluded at the index procedure (due to disease progression between the diagnostic angiogram and the study procedure). Another patient was excluded from final analysis because of inadvertent implantation of a non-study stent at the target segment. Therefore, 57 patients (38 in the SES and 19 in the BMS group) and 60 lesions (39 in the SES and 21 in the BMS group) were included for endpoint analysis.
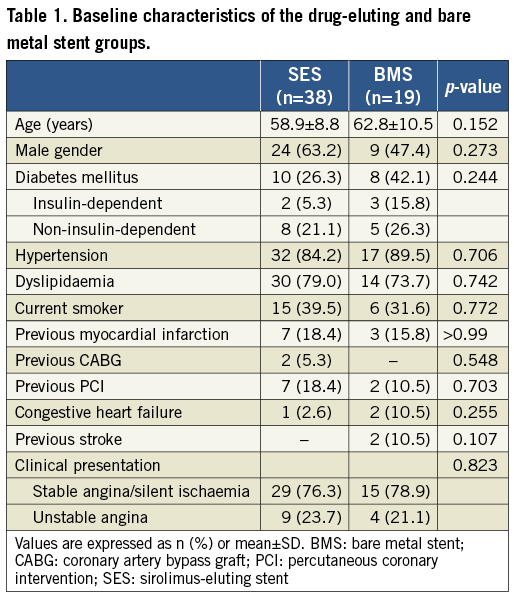
Patients’ characteristics did not reach statistically significant differences between the two groups (Table 1). Overall, the mean age was 60.2 ± 9.5 years and the majority were men (57.9%). Diabetes was present in 31.6% and 8.8% were insulin-dependent. Most patients had stable coronary disease (stable angina in 63.2% and silent ischaemia in 14.0%).
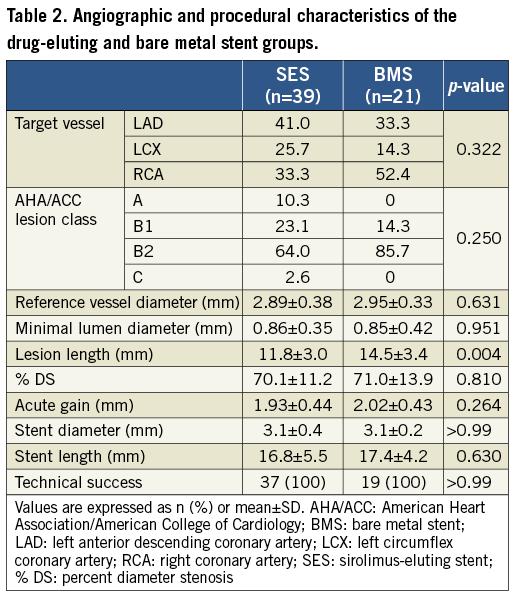
Procedural characteristics, presented in Table 2, were also similar between both groups. Right coronary artery PCI was performed in 40.0% of the patients, left anterior descending in 38.3%, and left circumflex in 21.7%. The mean vessel diameter was 2.9±0.37 mm and the mean minimal luminal diameter was 0.86±0.37 mm. The only difference between groups was the lesion length, shorter in the SES group (11.8±3.0 vs. 14.5±3.4; p=0.004). However, comparing the stent diameter and length between SES and BMS, they were similar in both groups.
QUANTITATIVE CORONARY ANGIOGRAPHY ANALYSIS AT SIX MONTHS
Angiographic follow-up, as shown in Table 3, was available at six months for all patients. The SES group showed a significant reduction of primary endpoint in-segment late loss (0.19±0.16 mm vs. 0.58±0.4 mm; p<0.001) (Figure 2). The SES arm also presented lower in-stent late loss, diameter of stenosis, and binary restenosis, as well as greater minimal luminal diameter as compared with the bare metal stent control group.
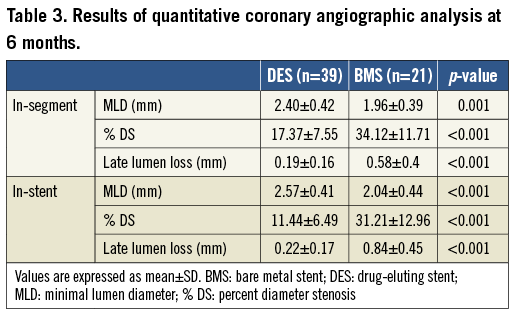
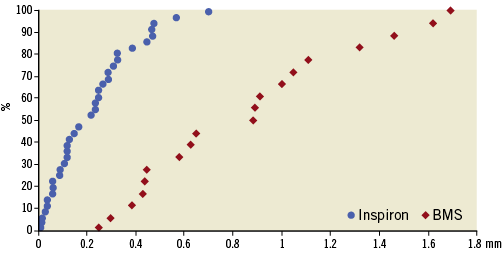
Figure 2. Cumulative frequency of in-segment angiographic late loss according to stent type at six months.
INTRAVASCULAR ULTRASOUND ANALYSIS
Intravascular data were available for 51 patients at six months. As demonstrated in Table 4 and Figure 3, the SES group had significantly lower hyperplasia volume compared to the BMS group (11.9±12.3 mm3 vs. 40.7±15.2 mm3, respectively; p<0.001) and there was significantly less percent neointimal obstruction (7.8±7.1% vs. 26.5±11.4%, respectively; p<0.001). The volume of incomplete stent apposition was similar between stents (0.05±0.32 mm3 vs. 0.46±1.67 mm3, respectively; p=0.18). There was no evidence of an “edge effect,” aneurysm formation, in-stent thrombosis or persistent dissection.
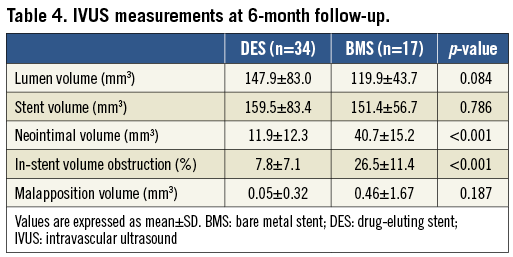
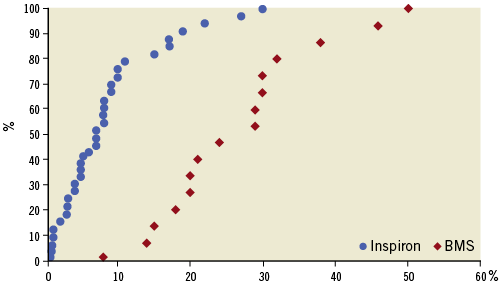
Figure 3. Cumulative frequency of stent volume of obstruction by intravascular ultrasound according to stent type at six months.
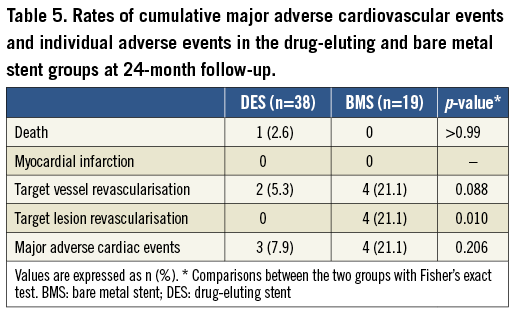
MAJOR ADVERSE CARDIAC EVENTS
Clinical follow-up up to 24 months was available in all 57 patients and is summarised in Table 5. Cumulative event rates were similar between groups. The cumulative incidence of MACE (death, myocardial infarction, and target vessel revascularisation) was 7.9% in the SES group versus 21.1% in the BMS group (p=0.2). There was one death in the SES arm due to an acute pulmonary oedema, without acute myocardial infarction. There were two target vessel revascularisations in the SES group but neither of them was a target lesion revascularisation. All four target vessel revascularisations in the bare metal group were in the segment of the index procedure. No acute myocardial infarction or stent thrombosis occurred in either group at the 24-month follow-up.
Discussion
The present first-in-man study tested a novel SES that combines a thin cobalt-chromium alloy platform with a bioabsorbable abluminal polymer, with a known efficacious drug to inhibit neointimal hyperplasia. This novel SES effectively suppressed neointimal growth at six months, as demonstrated angiographically and by IVUS.
Due to concern about the safety of first-generation DES, scientific interest has been aroused in developing newer devices, with improved polymer and metallic platform technologies. In experimental and clinical scenarios, thin-strut stents have been shown to produce less neointimal hyperplasia than thick-strut BMS6,7. Thick-strut (162 µm) stents have been shown to increase thrombogenicity as compared to identical thin-strut counterparts in ex vivo and in vivo models5. Moreover, in a mixed-treatment comparison analysis from 117,762 patient-years of follow-up from randomised trials, the thinner-strut cobalt-chromium everolimus-eluting stent was shown to be the safer, with significant reduction in myocardial infarction and stent thrombosis compared to BMS8. It is also known that local arterial injury due to durable polymers might be related to late and very late stent thrombosis3,4. The use of biodegradable polymer-coated stents may improve safety outcomes if the polymer and drug disappear after the full antirestenotic effect has been achieved, avoiding inflammatory side effects. Pooled individual patient data from three large-scale multicentre randomised clinical trials (ISAR-TEST 3, ISAR-TEST 4, and LEADERS 20) comparing biodegradable polymer DES with durable polymer SES have demonstrated that the first platform produced a lower risk of TVR (hazard ratio 0.82, 95% CI: 0.68-0.98, p=0.029) and stent thrombosis, (hazard ratio 0.56, 95% CI: 0.35-0.90, p=0.015), driven by a lower risk of very late stent thrombosis (hazard ratio 0.22, 95% CI: 0.08-0.61, p=0.004)9. Additionally, the design of exclusively abluminal coating allows lower drug concentration and might lead to even less vascular inflammation. Although the present study was powered for late loss and not for clinical events, we have shown a significant reduction in clinical restenosis driven by lower TLR up to two years, despite a similar MACE, and no stent thrombosis. Importantly, there were also no signs of local vascular toxicity and aneurysm formation at six-month imaging follow-up, and a similarly low malapposition volume was verified in the SES compared to the BMS group. The INSPIRON-I trial demonstrates the feasibility and early clinical safety and efficacy of this novel DES.
Limitations
The INSPIRON-I trial was designed to minimise the number of patients exposed to this novel device, while still providing adequate data about stent safety and efficacy. For this reason, the use of a BMS as comparator and in-segment LL as primary endpoint seemed appropriate but should be listed as a limitation. There are baseline clinical and angiographic statistically non-significant differences between the groups, but the small number of patients does not allow us to draw firm conclusions. Nonetheless, these data provide initial evidence for the proof of concept of this new DES platform and serve as a rationale to larger trials comparing this novel SES with other biodegradable polymer DES.
Conclusions
In conclusion, the Inspiron SES proved to be effective in reducing neointimal growth, leading to reduced target lesion revascularisation, with similar rates of death, myocardial infarction and stent thrombosis at two years.
| Impact on daily practice Drug-eluting stents have proven efficacy in reducing neointimal hyperplasia. However, factors such as thicker struts, the presence of a permanent polymer and antiproliferative drug may trigger vascular inflammatory reactions and thrombogenicity. The INSPIRON-I trial describes the first-in-man experience with a drug-eluting stent designed to address the aforementioned risks: thin cobalt-chromium alloy platform and bioabsorbable polymer eluting sirolimus from its abluminal surface. Compared with bare metal stents, the Inspiron sirolimus-eluting stent effectively suppressed neointimal growth at six months, as demonstrated angiographically and by IVUS. Associated with its efficacy, in this initial clinical experience, there was no evidence of safety concerns as assessed by clinical outcomes or imaging methods. |
Funding
Funded by Scitech Medical.
Conflict of interest statement
H.B. Ribeiro is supported by a PhD research grant from “CNPq, Conselho Nacional de Desenvolvimento Científico e Tecnológico - Brasil”. A.C. Lopes is supported in part by an Arie Research Fellowship from the Brazilian Society of Interventional Cardiology. The other authors have no conflicts of interest to declare.
