Abstract
Aims: Our aim was to investigate one-year outcomes in patients treated with bioresorbable scaffolds (BRS) for “off-label” versus currently “established” indications.
Methods and results: Consecutive patients treated with BRS between May 2012 and September 2014 in two centres were retrospectively recruited. Patients who met inclusion criteria as defined by the ABSORB III study were allocated to the established indication group (ESTG; 21 patients with 35 lesions) and the remaining patients to the off-label group (OFLG; 168 patients with 225 lesions). Target vessel failure (TVF) and ischaemia-driven target lesion revascularisation (id-TLR) at one year were evaluated in both groups. Patients in the OFLG had a higher prevalence of diabetes mellitus and longer lesion length. Predilatation, post-dilatation and intracoronary imaging were conducted in the majority of patients. At one-year follow-up, TVF (0% vs. 7.8%, p=0.32) and id-TLR (0% vs. 4.5%, p=0.31) occurred only in the OFLG with no adverse events in the ESTG. Definite stent thrombosis occurred in two OFLG patients (1.3%).
Conclusions: In a real-world setting, the majority (88.9%) of patients were treated with BRS for off-label indications. Off-label use of BRS appears to be associated with an acceptable occurrence of outcomes considering the greater complexity of this patient group.
Abbreviations
BRS: bioresorbable scaffold
DES: drug-eluting stent
ESTG: established indication group
id: ischaemia-driven
MI: myocardial infarction
OFLG: off-label indication group
TLR: target lesion revascularisation
TVF: target vessel failure
TVR: target vessel revascularisation
Introduction
Due to existing clinical trials restricting BRS use to “simple” lesions1,2 and limited use in routine clinical practice, only limited data from a few BRS registries which include these patients are available with limited follow-up3,4.
We investigated outcomes following BRS implantation for the treatment of complex “off-label” coronary lesions in comparison to “established” simple indications.
Methods
All patients who underwent implantation of the Absorb bioresorbable vascular scaffold (Abbott Vascular, Santa Clara, CA, USA) between May 2012 and September 2014 in two centres (San Raffaele Hospital and EMO-GVM Centro Cuore Columbus, Milan, Italy) were included in the present study. Patients who would have met inclusion criteria as defined by the ongoing ABSORB III trial5 (ClinicalTrials.gov number NCT01751906) (Online Appendix) were deemed to have been treated for an established indication and were allocated to the established use group (ESTG), whilst all other patients were assigned to the off-label group (OFLG).
The primary clinical outcome measure was target vessel failure (TVF) (composed of cardiac death, myocardial infarction [MI] excluding procedure-related MI, and ischaemia-driven target vessel revascularisation [id-TVR]) at one year. The secondary outcome measures were the occurrence of each component of TVF, ischaemia-driven target lesion revascularisation (id-TLR) and stent thrombosis at one year.
Results
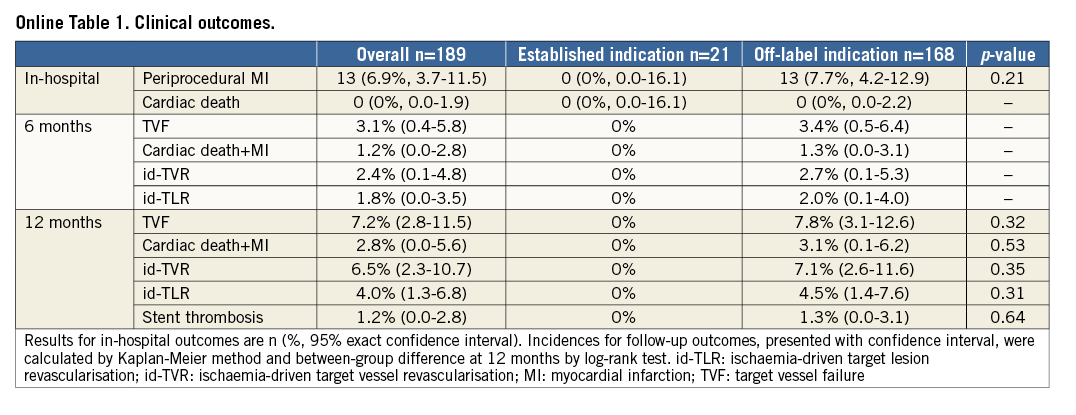
A total of 189 patients with 260 lesions underwent BRS implantation. Twenty-one patients (11.1%) with 35 lesions were allocated to the ESTG and 168 patients (88.9%) with 225 lesions to the OFLG. The median follow-up period was 393 days (interquartile range: 208-549 days). Patients in the OFLG demonstrated a higher prevalence of diabetes mellitus (ESTG vs. OFLG: 4.8% vs. 28.6%, p=0.031) and a higher SYNTAX score (11.5±9.1 vs. 16.9±9.1, p=0.013) (Table 1).
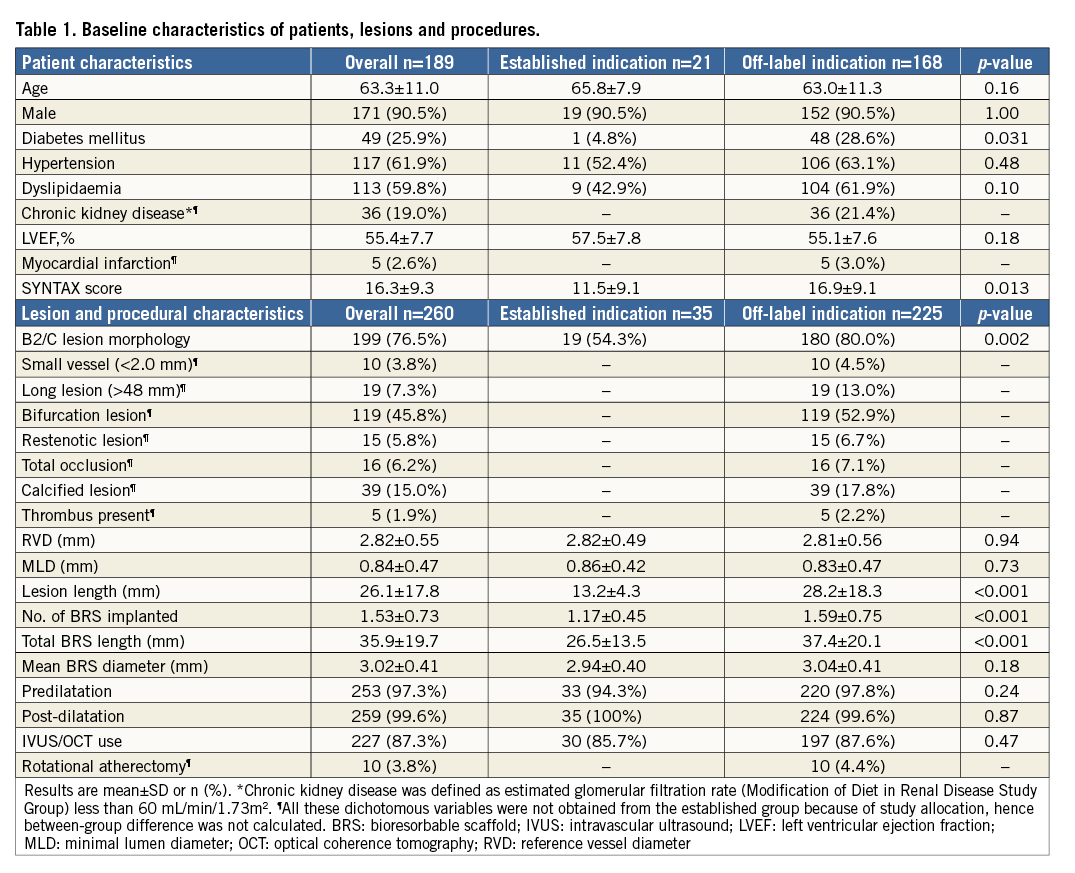
With regard to the whole study population, there was a high prevalence of bifurcation involvement (45.8%), long lesions (36.0±25.3 mm) and BRS length (49.4±30.1 mm). As expected, lesion length (13.2±4.3 mm vs. 28.2±18.3 mm, p<0.001) and length of implanted scaffold (26.5±13.5 mm vs. 37.4±20.1 mm, p<0.001) were significantly greater in the OFLG. Both reference vessel diameter (p=0.94) and scaffold diameter (p=0.73) were similar between the two groups (Table 1).
Predilatation (97.3%), post-dilatation (99.6%) and intracoronary imaging (87.3%) were performed in the majority of patients in both groups (Table 1).
Clinical outcomes
At one-year follow-up, all events including TVF (0% vs. 7.8%, p=0.32) and id-TLR (0% vs. 4.5%, p=0.31) occurred only in the OFLG (Figure 1, Online Table 1). With regard to in-hospital events, 13 cases (6.9% of the overall cohort) of periprocedural MI were observed in the OFLG (7.7% of the OFLG, between-group difference: p=0.21) with no events in the ESTG (Online Table 1). Definite stent thrombosis occurred in two OFLG patients, the first two hours following primary percutaneous coronary intervention for ST-elevation MI, and the second 149 days following BRS implantation in the context of premature discontinuation of dual antiplatelet therapy. Therefore, the incidence of all stent thrombosis at one year was 1.2% for the overall cohort and 1.3% for the off-label group (vs. ESTG [0%], p=0.64). Both patients underwent successful emergency treatment with the implantation of second-generation drug-eluting stents (DES).
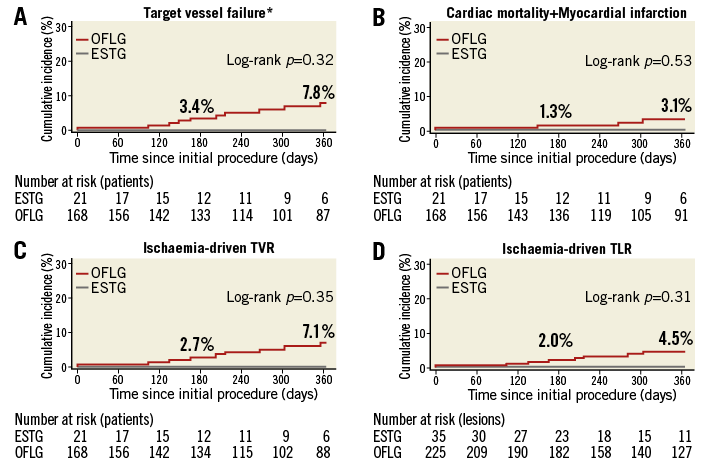
Figure 1. Kaplan-Meier cumulative event curves for the primary and secondary endpoints at two years. *Target vessel failure is a composite of cardiac mortality, any myocardial infarction other than periprocedural, and target vessel revascularisation. ESTG: established indication group; OFLG: off-label indication group; TLR: target lesion revascularisation; TVR: target vessel revascularisation
Discussion
The principal findings of this study are: i) the majority of patients who underwent BRS implantation in this real-world patient group did not present with simple lesions; ii) the patients and coronary lesions were of greater complexity and patients had a higher risk when compared to those in the prior ABSORB trials2,6; and iii) the occurrence of each clinical outcome measure was acceptable.
The Absorb BRS has demonstrated promising clinical results comparable to new-generation metallic stents in the initial trials such as ABSORB EXTEND6. In the current study, there was a high prevalence of diabetes mellitus, chronic kidney disease, B2/C lesions, bifurcations, and longer lesion length, when compared to the GHOST-EU (Gauging coronary Healing with biOresorbable Scaffolding plaTforms in EUrope) registry (TVF: 4.9%, cardiac death: 1.0%, MI: 2.7%, TLR: 2.5% at six months)3, yet the patients in the OFLG showed comparable six-month clinical outcomes. Additionally, one-year clinical outcomes of the OFLG were similar to the six-month outcomes demonstrated in the Dutch AMC (Academic Medical Center) registry (TVF: 8.5%, cardiac death: 0.8%, MI: 3.0%, TLR: 6.3%)4. These data may help to support the use of BRS in higher-risk patients with more complex lesions if optimal implantation techniques are utilised. Of note, we found that no adverse events occurred in the ESTG during one-year follow-up, supporting the use of BRS in this patient group.
Currently, in clinical practice only 10% of patients meet the established indication for BRS implantation, with few data with regard to more complex disease. High-risk patients who present with more complex disease, including long segments of diffuse disease, bifurcations requiring side branch jailing or in-stent restenosis resulting in multilayer struts, may benefit from the advantages associated with this innovative device. In this study, predilatation, post-dilatation and IVUS guidance were performed in the majority of patients, in keeping with the recommendations of a recent expert consensus report7, and this may have contributed to better outcomes in this cohort.
Whilst the outcomes we report appear to be superior to those from other currently available BRS registries, it is important to note that they are still inferior to equivalent DES. The reasons for this disparity need to be investigated in future studies, with a possible explanation being the greater strut width and thickness of current BRS devices.
Limitations
The current study was retrospective and patients were not randomised. All decisions with regard to the indication, procedural strategy and use of BRS were left to the operator’s discretion. Given the small sample size, the study was not adequately powered to detect differences in clinical outcomes.
Conclusions
In a real-world patient population, 88.9% of patients were treated with BRS for off-label indications. Our findings may support the use of BRS in off-label indications, provided optimal implantation techniques are utilised.
| Impact on daily practice Although there is an increasing trend to expand the use of BRS for indications that have not been investigated in large studies, it is important that a number of practical aspects are carefully considered. Bearing in mind the reduced radial force and tensile strength in comparison to contemporary metallic DES, meticulous lesion preparation and post-dilatation with utilisation of intracoronary imaging may be important to obtain optimal clinical outcomes and to take full advantage of the benefits of this novel device. |
Conflict of interest statement
A. Latib is a consultant for Medtronic. The other authors have no conflicts of interest to declare.
SUPPLEMENTARY DATA
Online Appendix. Inclusion criteria preliminarily established for the ABSORB III study
(ClinicalTrials.gov Identifier: NCT01751906)
GENERAL INCLUSION CRITERIA
18 years of age.
Subject or a legally authorised representative must provide written informed Consent prior to any study related procedure, per site requirements.
Subject must have evidence of myocardial ischaemia. In the absence of noninvasive ischaemia, fractional flow reserve (FFR) must be done and indicative of ischaemia.
Acceptable candidate for coronary artery bypass graft (CABG) surgery.
Female subject of childbearing potential who does not plan pregnancy for up to 1 year following the index procedure. For a female subject of childbearing potential a pregnancy test must be performed with negative results known within 7 days prior to the index procedure per site standard.
Female subject is not breast-feeding at the time of the screening visit and will not be breast-feeding for up to 1 year following the index procedure.
Subject agrees to not participate in any other investigational or invasive clinical study for a period of 1 year following the index procedure.
GENERAL EXCLUSION CRITERIA
Any surgery requiring general anesthesia or discontinuation of aspirin and/or an adenosine diphosphate (ADP) antagonist is planned within 12 months after the procedure.
Hypersensitivity or contraindication to device material and its degradants (everolimus, poly (L-lactide), poly (DL-lactide), lactide, lactic acid) and cobalt, chromium, nickel, platinum, tungsten, acrylic and fluoro polymers that cannot be adequately pre-medicated. Subject has a known contrast sensitivity that cannot be adequately pre-medicated.
Allergic reaction, hypersensitivity or contraindication to aspirin; or to clopidogrel and prasugrel and ticagrelor; or to heparin and bivalirudin, and therefore cannot be adequately treated with study medications.
Acute myocardial infarction (AMI: STEMI or NSTEMI) within 72 hours of the index procedure and both creatine kinase (CK) and CK-MB have not returned to within normal limits at the time of index procedure; or subject with stable angina or silent ischaemia has CK-MB that is greater than normal limits at the time of the index procedure.
Subject is currently experiencing clinical symptoms consistent with new onset AMI (STEMI or NSTEMI), such as nitrate-unresponsive prolonged chest pain with ischemic ECG changes.
Cardiac arrhythmia as identified at the time of screening for which at least one of the following criteria is met:
a. Subject requires coumadin or any other agent for chronic oral anticoagulation
b. Subject is likely to become hemodynamically unstable due to their arrhythmia
c. Subject has poor survival prognosis due to their arrhythmia
Left ventricular ejection fraction (LVEF) <30%.
Subject has undergone prior percutaneous coronary intervention (PCI) within the target vessel during the last 12 months. Prior PCI within the non-target vessel or any peripheral intervention is acceptable if performed anytime >30 days before the index procedure, or between 24 hours and 30 days before the index procedure if successful and uncomplicated.
Future staged PCI either in target or non-target vessels or subject requires future peripheral interventions <30 days after the index procedure.
Subject has received any solid organ transplants or is on a waiting list for any solid organ transplants.
At the time of screening, the subject has a malignancy that is not in remission.
Subject is receiving immunosuppressant therapy or has known immunosuppressive or severe autoimmune disease that requires chronic immunosuppressive therapy (e.g., human immunodeficiency virus, systemic lupus erythematosus, etc.). Note: corticosteroids are not included as immunosuppressant therapy.
Subject has previously received or is scheduled to receive radiotherapy to a coronary artery (vascular brachytherapy), or the chest/mediastinum.
Subject is receiving or will require chronic anticoagulation therapy (e.g., coumadin, dabigatran, apixaban, rivaroxaban or any other agent for any reason).
Subject has a platelet count <100,000 cells/mm3 or >700,000 cells/mm3.
Subject has a documented or suspected hepatic disorder as defined as cirrhosis or Child-Pugh ≥Class B.
Renal insufficiency. NOTE: Estimated glomerular filtration rate (GFR) can be based on Modification of Diet in Renal Disease (MDRD) equation or Cockcroft-Gault equation (CCG).
High risk of bleeding for any reason; has a history of bleeding diathesis or coagulopathy; has had a significant gastro-intestinal or significant urinary bleed within the past six months.
Cerebrovascular accident or transient ischemic neurological attack (TIA) within the past six months, or any prior intracranial bleed, or any permanent neurologic defect, or any known intracranial pathology (e.g., aneurysm, arteriovenous malformation, etc.).
Extensive peripheral vascular disease that precludes safe 6 French sheath insertion. Note: femoral arterial disease does not exclude the patient if radial access may be used.
Subject has life expectancy <5 years for any non-cardiac cause or cardiac cause.
Subject is in the opinion of the Investigator or designee, unable to comply with the requirements of the study protocol or is unsuitable for the study for any reason. This includes completion of Patient Reported Outcome instruments.
Subject is currently participating in another clinical trial that has not yet completed its primary endpoint.
Vulnerable population.
ANGIOGRAPHIC INCLUSION CRITERIA
1. One or two de novo target lesions:
a. If there is one target lesion, a second non-target lesion may be treated but the non-target lesion must be present in a different epicardial vessel, and must be treated first with a successful, uncomplicated result prior to randomisation of the target lesion.
b. If two target lesions are present, they must be present in different epicardial vessels and both must satisfy the angiographic eligibility criteria.
c. The definition of epicardial vessels means the LAD, LCX and RCA and their branches. Thus, the patient must not have lesions requiring treatment in e.g. both the LAD and a diagonal branch.
2. Target lesion(s) must be located in a native coronary artery with a visually estimated or quantitatively assessed % diameter stenosis (DS) of ≥50% and <100% with a thrombolysis in myocardial infarction (TIMI) flow of ≥1 and one of the following: stenosis ≥70%, an abnormal functional test (e.g. fractional flow reserve, stress test), unstable angina or post-infarct angina.
a. Lesion(s) must be located in a native coronary artery with RVD by visual estimation of ≥2.5 mm and ≤3.75 mm.
b. Lesion(s) must be located in a native coronary artery with length by visual estimation of ≤24 mm.
c. For Lead-in subjects with 3.0×18 mm Absorb BVS: lesion(s) must be located in a native coronary artery with RVD by visual estimation of ≥2.75 mm and ≤3.25 mm. The lesion length by visual estimation is ≥8 mm and ≤14 mm.
ANGIOGRAPHIC EXCLUSION CRITERIA
All exclusion criteria apply to the target lesion(s) or target vessel(s).
1. Lesion which prevents successful balloon pre-dilatation, defined as full balloon expansion with the following outcomes:
a. Residual %DS is a maximum of <40% (per visual estimation), ≤20% is strongly recommended.
b. TIMI Grade-3 flow (per visual estimation).
c. No angiographic complications (e.g. distal embolisation, side branch closure).
d. No dissections National Heart Lung and Blood Institute (NHLBI) grade D-F.
e. No chest pain lasting >5 minutes.
f. No ST depression or elevation lasting >5 minutes
2. Lesion is located in left main.
3. Aorto-ostial right coronary artery (RCA) lesion (within 3 mm of the ostium).
4. Lesion located within 3 mm of the origin of the Left Anterior Descending Artery (LAD) or left circumflex artery (LCX).
5. Lesion involving a bifurcation with a:
a. side branch ≥2 mm in diameter, or
b. side branch with either an ostial or non-ostial lesion with diameter stenosis >50%, or
c. side branch requiring dilatation.
6. Anatomy proximal to or within the lesion that may impair delivery of the Absorb BVS or XIENCE stent:
a. Extreme angulation (≥90°) proximal to or within the target lesion.
b. Excessive tortuosity (≥two 45° angles) proximal to or within the target lesion.
c. Moderate or heavy calcification proximal to or within the target lesion. If IVUS used, subject must be excluded if calcium arc in the vessel prior to the lesion or within the lesion is ≥180°.
7. Vessel contains thrombus as indicated in the angiographic images or by IVUS or OCT.
8. Lesion or vessel involves a myocardial bridge.
9. Vessel has been previously treated with a stent at any time prior to the index procedure such that the Absorb BVS or XIENCE would need to cross the stent to reach the target lesion.
10. Vessel has been previously treated and the target lesion is within 5 mm proximal or distal to a previously treated lesion.
11. Target lesion located within an arterial or saphenous vein graft or distal to any arterial or saphenous vein graft.