Abstract
Background: The multicentre, randomised, sham-controlled RADIANCE-HTN SOLO trial reported the blood pressure (BP)-lowering efficacy and safety of ultrasound renal denervation (RDN) in the absence (2 months) and presence (6 and 12 months) of antihypertensive medications in patients with mild-to-moderate hypertension.
Aims: The aim of this report was to evaluate patients originally assigned to the sham group who crossed over to RDN.
Methods: After the primary endpoint was met, patients in the sham arm who remained uncontrolled were allowed to cross over to receive RDN. All patients were unblinded and treated with standard of care medications at the time of crossover. Ambulatory BP was evaluated 2 and 6 months after crossover.
Results: Among 72 subjects of the sham arm, 33 underwent ultrasound RDN after an average follow-up of 23±6 months. Prior to crossover, patients had a daytime ambulatory BP of 144.1±10.1/89.9±8.4 mmHg and received 1.2±0.8 antihypertensive medications. Mean change in daytime ambulatory BP from pre-crossover to 2 and 6 months post RDN was –11.2±13.7/–7.1±8.9 mmHg (n=33; p<0.001; p<0.001) and –10.8±17.3/–7.8±11.6 mmHg (n=27; p=0.002; p<0.001). The number of antihypertensive medications did not change from pre-crossover baseline to 2 and 6 months. Eighteen of 33 (54.5%) patients had their daytime ambulatory BP controlled (<135/85 mmHg) at 2 months and 44.4% (12/27) at 6 months post RDN. No major procedure-related adverse events occurred.
Conclusions: During unblinded long-term follow-up of the RADIANCE-HTN SOLO study, patients originally assigned to a sham procedure who remained uncontrolled had significant reductions in BP following crossover treatment with ultrasound RDN.
Introduction
Hypertension is the leading risk factor for global disease burden and is strongly associated with cardiovascular morbidity and with mortality1. Despite the availability of multiple classes of effective antihypertensive medication, hypertension remains uncontrolled in a large percentage of patients2.
Activation of the sympathetic nervous system contributes to the development and maintenance of hypertension and is associated with comorbidities3. Increased systemic sympathetic activity, especially of the renal sympathetic nervous system, is thought to play a key role in the pathogenesis and perpetuation of hypertension4. Catheter-based renal denervation (RDN), with either ultrasound, radiofrequency, or injection of alcohol, represents an alternative treatment approach for uncontrolled hypertension5.
The RADIANCE-HTN trial programme was designed to compare the blood pressure (BP)-lowering efficacy and safety of endovascular ultrasound RDN with a sham procedure6. The results of the international, multicentre, RADIANCE-HTN SOLO trial were reported previously7,8,9. The trial met its primary endpoint, demonstrating a reduction in daytime ambulatory systolic BP (dASBP) of 6.3 mmHg (95% CI: –9.4 to –3.1, p=0.0001) with RDN relative to a sham procedure at 2 months with patients maintained off medications7. BP reductions were sustained in the RDN arm at 6- and 12-month follow-up with decreased medication burden as compared to sham8,9. As pre-specified in the original study design, after the primary endpoint was met, patients of the sham group who remained with uncontrolled hypertension were permitted to receive RDN (designated as “crossover”).
The initial RADIANCE-HTN SOLO results were in patients on no medications at 2 months and on a standardised stepped care uptitration of medication at 6 months. Since crossover patients were treated or untreated at the time of the crossover procedure and throughout follow-up at the discretion of the investigator, analysis of the results of this crossover cohort allows a unique understanding of the efficacy and safety of RDN in patients already receiving antihypertensive therapy. This analysis represents data on the cohort of patients who had crossed over and received RDN as of February 2020.
Methods
STUDY DESIGN AND STUDY POPULATION
The study design and primary results of the RADIANCE-HTN SOLO trial have been described in detail previously6,7,8. The study was approved by local ethics committees or institutional review boards and conducted in accordance with the Declaration of Helsinki. All patients provided written informed consent. Between March 2016 and December 2017, participants were recruited into the RADIANCE-HTN SOLO trial from 21 centres in the USA and 18 in Europe (located in France, Germany, the Netherlands, Belgium, and the United Kingdom).
In brief, RADIANCE-HTN SOLO is an international, multicentre, randomised, sham-controlled trial of 146 patients that compared changes in BP with endovascular ultrasound RDN with the Paradise™ RDN system (ReCor Medical, Inc., Palo Alto, CA, USA) to a sham procedure consisting of a renal angiogram only. Key inclusion criteria were uncontrolled hypertension with office BP ≥140/90 mmHg and <180/110 mmHg on 0-2 antihypertensive medications or controlled hypertension with office BP <140/90 mmHg on 1 or 2 antihypertensive medications, estimated glomerular filtration rate (eGFR) of ≥40 mL/min/1.73 m² (based on the Modification of Diet in Renal Disease formula) and no history of cardiovascular or cerebrovascular events. Patients who met these initial inclusion criteria were taken off all antihypertensive medications and qualified for randomisation if mean daytime ambulatory BP was ≥135 mmHg systolic and ≥85 mmHg diastolic and renal anatomy was considered appropriate for the procedure.
Patients were to remain off antihypertensive medications throughout the first 2 months of follow-up unless safety BP criteria were exceeded. Between 2 and 5 months, if monthly measured home BP was ≥135/85 mmHg, a standardised stepped-care antihypertensive treatment was recommended, as previously described6. After the 6-month efficacy and safety endpoints were ascertained, patients and clinical staff were unblinded to group assignment (RDN or sham). After 6 months, antihypertensive therapy was prescribed at the treating physician’s discretion with medication initiation or titration and clinical visits per standard of care.
After the primary endpoint was met, unblinded crossover for patients initially randomised to the sham procedure was permitted if specific criteria were met. Namely, patients had to have a re-qualifying dASBP ≥135 and/or dADBP ≥85 mmHg, suitable renal anatomy, and meet all the inclusion/exclusion criteria, as previously described. The re-qualifying ambulatory BP measurement was used as the “pre-crossover baseline” reading.
In contrast to the primary part of the study, there was no protocol-defined antihypertensive medication titration protocol following the crossover procedure. Antihypertensive medications could be adjusted as needed at the investigator’s discretion.
The methods to measure office BP (Omron M10-IT; Omron Healthcare, Kyoto, Japan) and ABPM (Microlife WatchBP, Taipei, Taiwan) have been reported previously7.
OUTCOMES
The primary analysis was the mean change in dASBP from pre-crossover baseline to 2 months and 6 months. Medication burden was assessed by total number of antihypertensive medications (regardless of the dose) and the sum of defined daily dose (DDD) of each individual antihypertensive medication10. Additional analyses performed were changes in average 24-hr ambulatory, night-time ambulatory, and office BP. The proportion of patients with controlled dASBP (<135/85 mmHg) and controlled office BP (<140/90 mmHg), proportion of responders (defined as a ≥5 mmHg decrease in dASBP) and change in eGFR at 2 and 6 months were also assessed.
Safety assessments were performed as previously reported6,7,8. All pre-specified potential device or procedural and/or serious adverse events reported by study sites were sent for independent adjudication. An independent data safety and monitoring board reviewed study data quarterly for all enrolled patients.
Major adverse events were defined as all-cause mortality, renal failure (eGFR <15 mL/min/1.73 m2 or need for renal replacement therapy or doubling of serum creatinine), embolic event resulting in end-organ damage, renal artery, or other major vascular complications requiring intervention, and hospitalisation for hypertensive crisis within 30 days or new renal artery stenosis greater than 70% by duplex ultrasound and confirmed by renal computed tomography (CT) or magnetic resonance (MR) angiography within 6 months.
STATISTICAL ANALYSIS
Continuous variables are expressed as mean±standard deviation. The signed-rank test was used to compare BP values and medications between time points. All analyses were performed using Statistical Analysis System (SAS) software, version 9.4 (SAS Institute, Cary, NC, USA). A p-value lower than 0.05 (two-sided) was considered significant.
Results
STUDY POPULATION
Patients were maintained in their initial assigned group with no crossover until the primary endpoint was met. Of the 72 patients initially allocated to the sham group, 2 were lost to follow-up and 1 died prior to 12 months. Of the remaining 69 patients, 33 crossed over and received ultrasound RDN and were included in this analysis. There were 3 additional patients who crossed over after the cut-off date for this analysis and whose follow-up is ongoing but not complete. There were 33 patients who did not cross over (11 did not meet the crossover criteria, 13 chose not to cross over by either patient or physician decision, 4 are pending crossover, 4 were lost to follow-up, and 1 withdrew consent) (Figure 1). The mean time from randomisation to crossover was 23±6 months and the mean time between pre-crossover ABPM and the crossover procedure was 22±15 days.
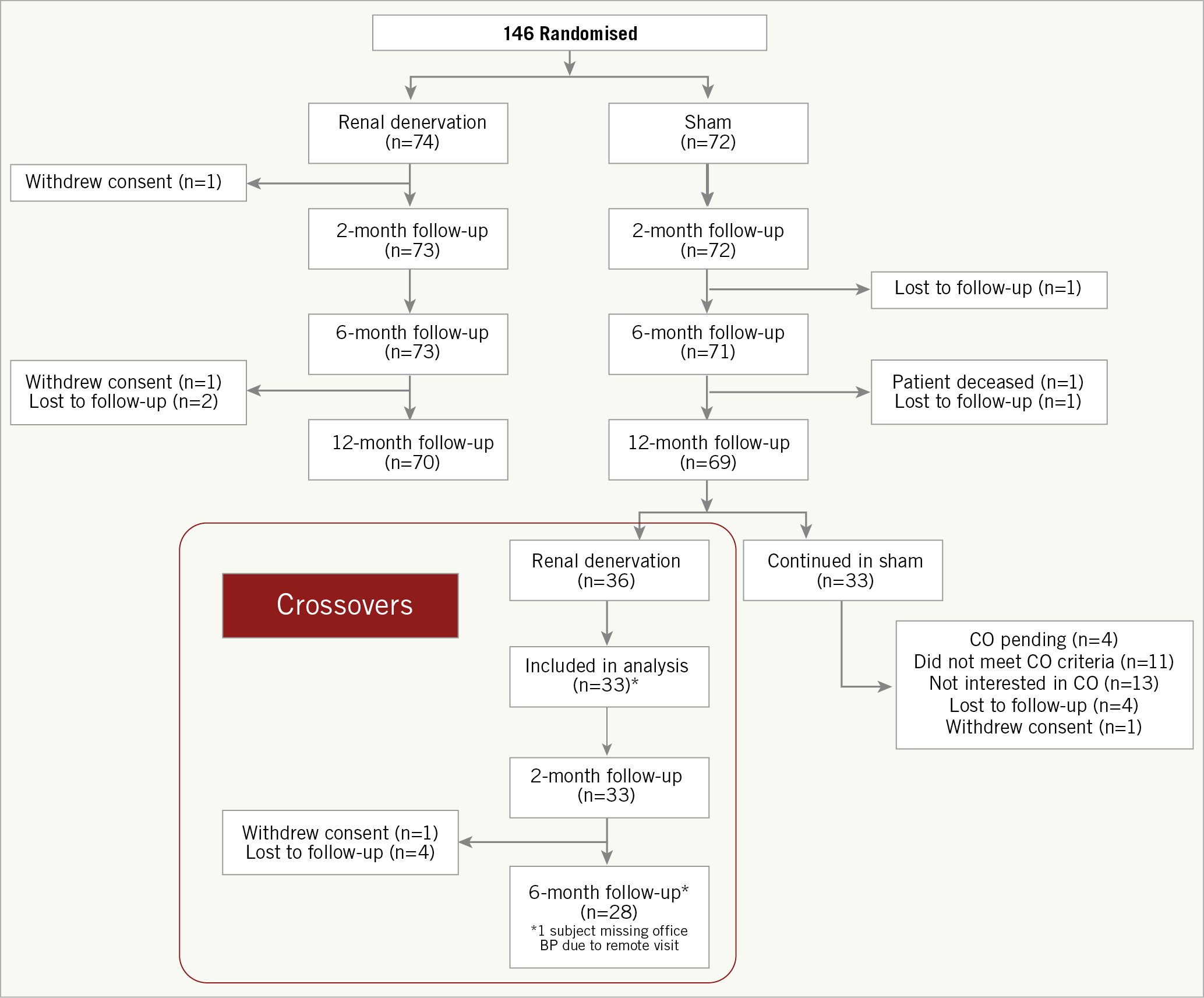
Figure 1. Patient disposition. *Three patients crossed over after the cut-off date for this analysis. CO: crossover
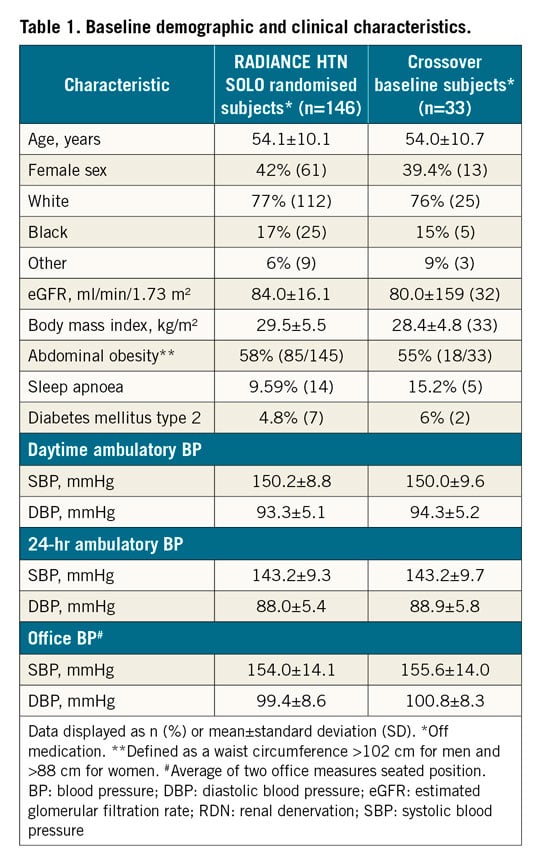
Baseline characteristics were similar between the overall SOLO cohort and the crossover cohort at baseline pre-randomisation (Table 1). In the crossover group, the mean age was 54 years, 39% were female and 76% were white, 55% had abdominal obesity and 15% reported sleep apnoea. The average number of antihypertensive medications at the time of crossover was 1.2±0.8 (Supplementary Table 1). At the time of crossover, 7 (21.2%) patients were not receiving any antihypertensive medications, 14 (42.4%) were receiving one medication, and 11 (33.3%) were receiving two medications.
Crossover patients received an average of 5.8±0.7 ultrasound emissions with an average procedure time of 72 minutes that was similar to that reported in the initial cohort. An average of 142±63 cm3 of contrast was used per patient (Supplementary Table 2).
OUTCOMES
24-hr ABPM was available in 33 patients at 2 months and 27 patients at 6 months (Figure 1). The average decrease in daytime systolic and diastolic ambulatory BP at 2 months from pre-crossover baseline was 11.2±13.7 mmHg systolic (p<0.001) and 7.1±8.9 mmHg diastolic (p<0.001) (Figure 2, Table 2). At 6 months, the average reduction from crossover in dASBP was 10.8±17.3 mmHg (p=0.002) and in dADBP was 7.8±11.6 mmHg (p<0.001) (Figure 2, Table 2). Individual patient changes in dASBP are shown in Supplementary Figure 1.
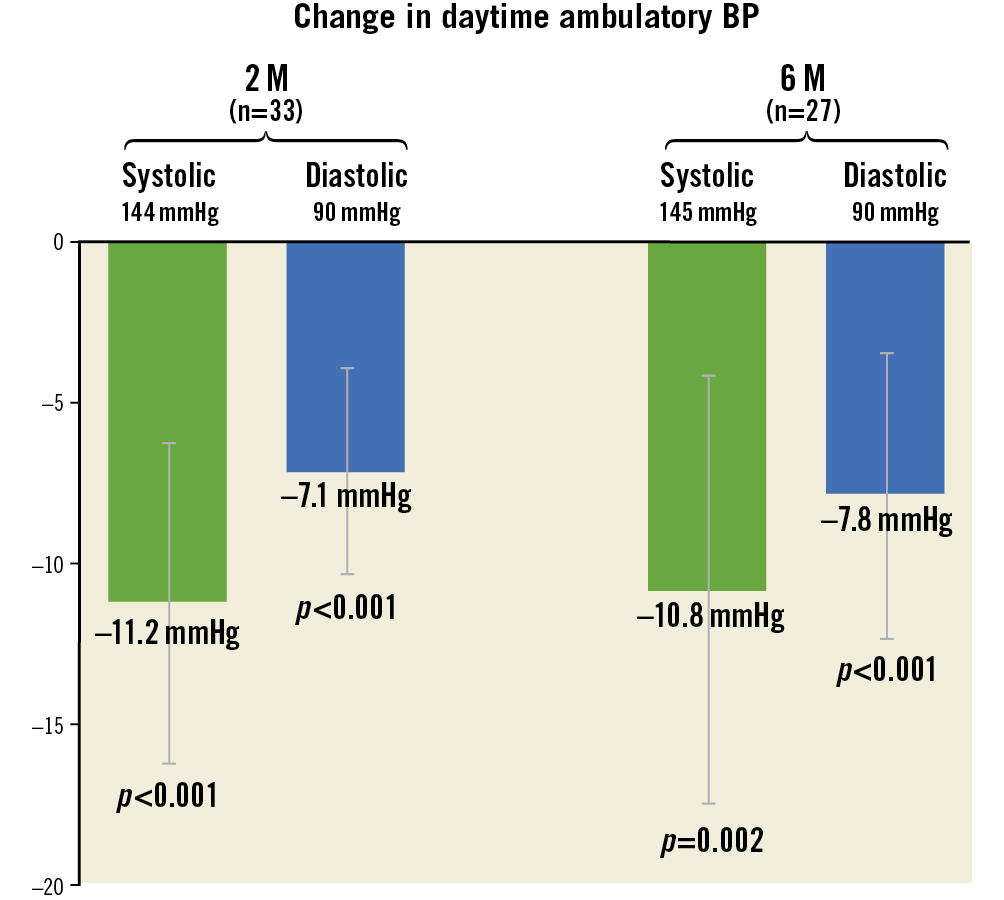
Figure 2. Change in daytime ambulatory blood pressure from baseline to 2 and 6 months. Data presented as mean and 95% CIs; p-value for change from baseline is from signed-rank test; matched 2- and 6-month data.
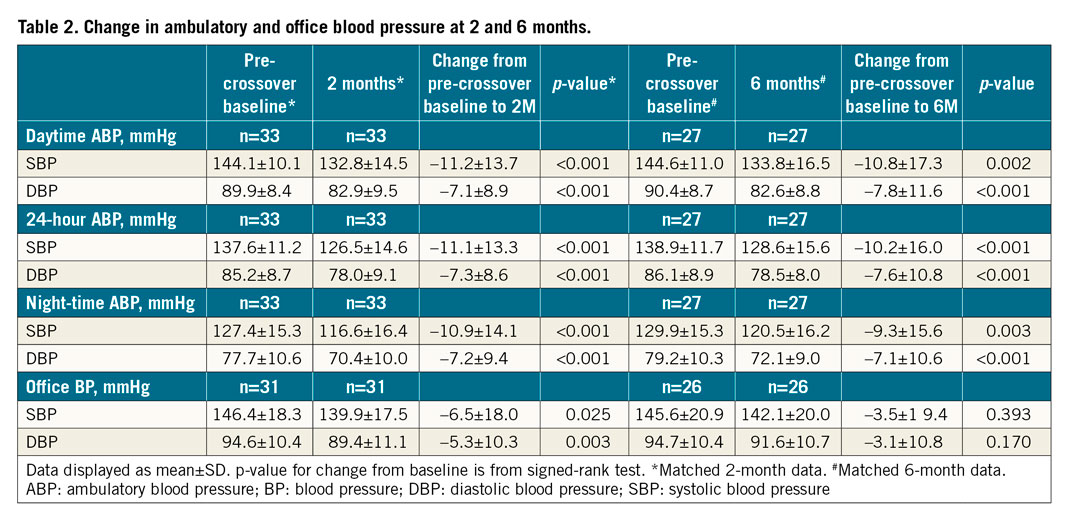
Decreases in 24-hr, daytime and night-time ambulatory systolic BP and diastolic BP were also significant and comparable to daytime ambulatory BP at 2 and 6 months (Table 2, Figure 3). There was no difference in 24-hr, daytime and night-time ambulatory systolic BP and diastolic BP between 2 and 6 months (24-hr SBP: p=0.371, DBP: p=0.566; daytime SBP: p=0.59, DBP: p=0.657; night-time SBP: p=0.171, DBP: p=0.192). Additionally, there was a significant decrease in office systolic and diastolic BP at 2 months and a trend towards a decrease at 6 months (Table 2).
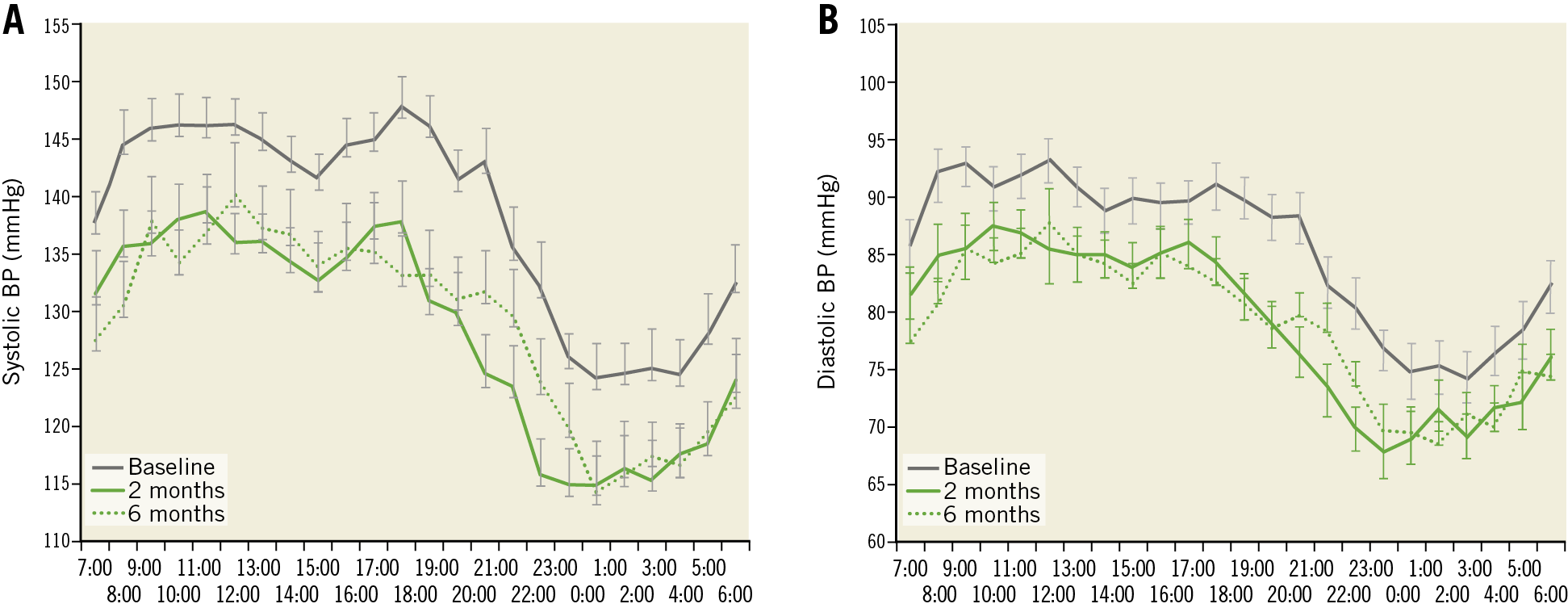
Figure 3. 24-hour ambulatory profiles at baseline at crossover and 2 and 6 months post crossover. A) Systolic blood pressure. B) Diastolic blood pressure. Error bars represent standard error. BP: blood pressure
After crossover, daytime ambulatory BP was controlled (<135/85 mmHg) in 54.5% (18/33) at 2 months (p<0.001) and 44.4% (12/27) at 6 months (p=0.002) (Figure 4). Office BP was controlled (<140/90 mmHg) in 41.9% (13/31) at 2 months and 23.1% (6/26) at 6 months.
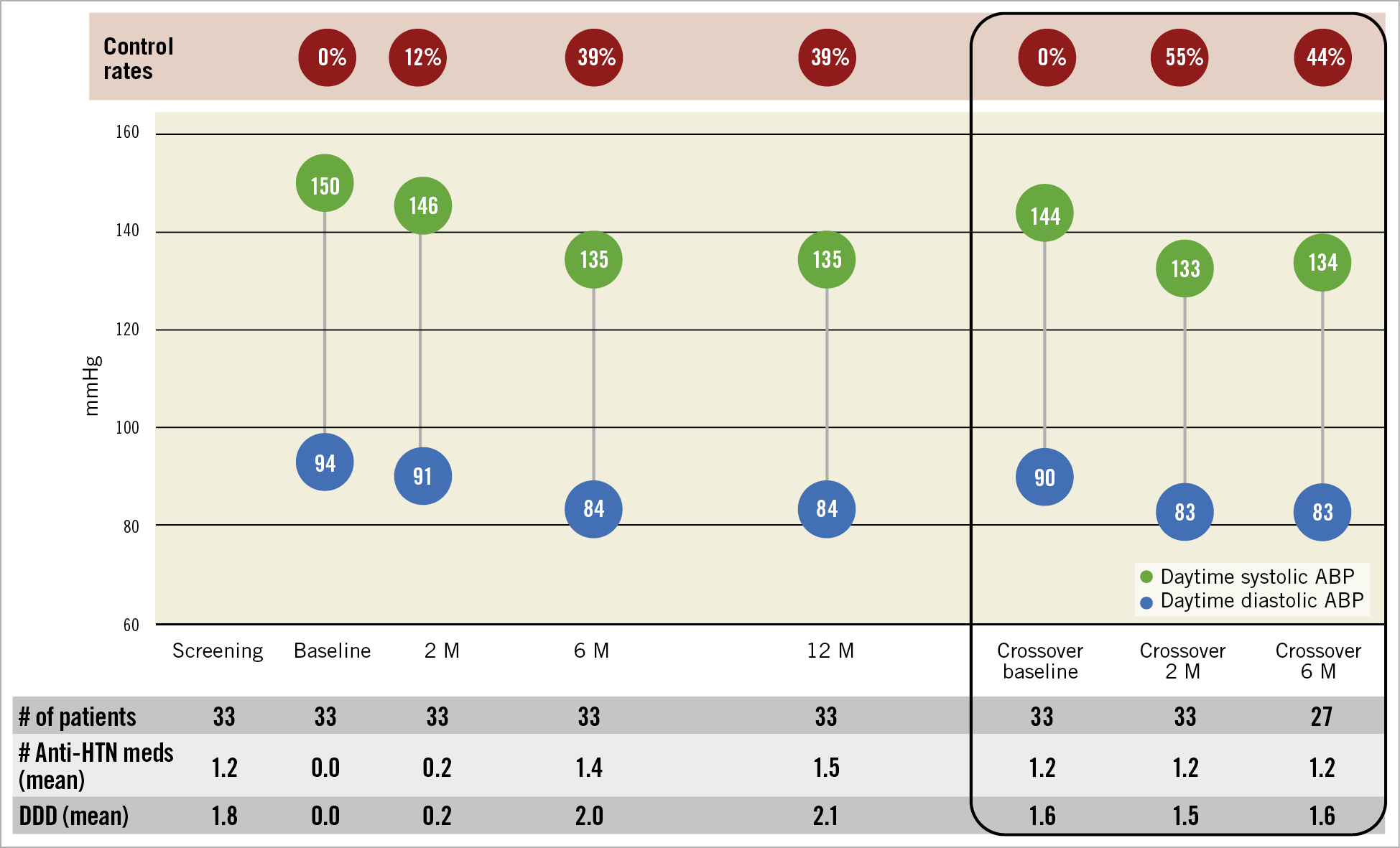
Figure 4. Daytime systolic and diastolic ambulatory BP, number of antihypertensive medications and defined daily dose. Control defined as BP <135/85 mmHg. ABP: ambulatory blood pressure; DDD: defined daily dose; HTN: hypertension
The percentage of patients with at least a 5 mmHg reduction in dASBP was 72.7% (24/33) at 2 months and 62.9% (17/27) at 6 months.
At 2 and 6 months, 78.8% (26/33) and 74.1% (20/27) of patients were receiving antihypertensive medications. The total number of antihypertensive medications (Figure 4) and DDD did not change significantly between baseline, 2 and 6 months (Supplementary Table 3).
SAFETY OUTCOMES
There were no major adverse events (Supplementary Table 4). There was one episode of pulmonary embolism 86 days post crossover procedure that was unrelated to the procedure. eGFR remained unchanged up to 6 months after RDN (Supplementary Table 5). There was no evidence of new or worsening renal artery stenosis or other vascular events reported up to the 6-month follow-up.
Discussion
The findings of this post hoc analysis of the RADIANCE-HTN SOLO trial indicate that at 2 and 6 months post-crossover to RDN, dASBP decreased by an average of approximately 11 mmHg, while medication burden remained unchanged in patients with mild to moderate hypertension initially randomised to the sham group and who remained uncontrolled despite add-on medications (Central illustration). There was no amplification or reduction of the magnitude of the average BP decrease between 2 and 6 months. There was one episode of pulmonary embolism that was unrelated to the procedure post RDN.
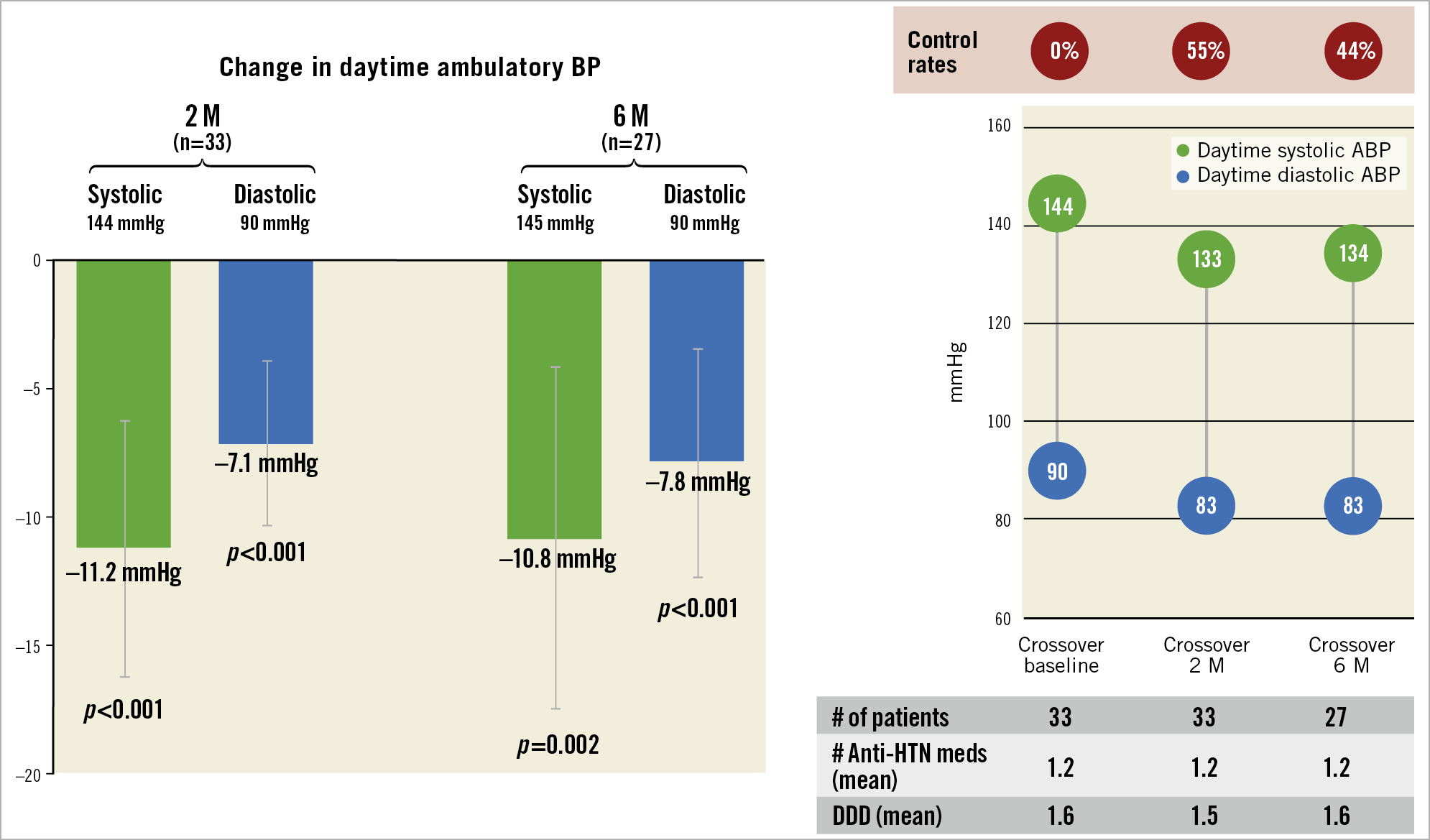
Central illustration. Medication burden and 24-hr ambulatory blood pressure profiles at 12 months in the RADIANCE-HTN SOLO trial. ABP: ambulatory blood pressure; DDD: defined daily dose; HTN: hypertension
The results of the present analysis are consistent with the primary outcome of the RADIANCE-HTN SOLO trial. However, there was no strict drug titration protocol applied between 2 and 6 months for crossover patients, so this may provide additional insights into the efficacy and 6-month durability of ultrasound RDN when used under less strict conditions. In the 6-month analysis of the RADIANCE-HTN SOLO trial8, patients treated with RDN had a greater reduction in dASBP when compared with sham (difference adjusted for baseline BP and number of medications: –4.3 mmHg; 95% CI: –7.9 to –0.6; p=0.024) although they had a lower medication burden. Following the collection of the 6-month data, patients and investigators were unblinded and the antihypertensive medication could be modified at the physician’s discretion. Importantly, the BP-lowering effect of RDN was sustained at 12 months (-16.5±12.9 mmHg in dASBP9) and, as compared with the sham group, patients treated with RDN received fewer antihypertensive drugs (1.0 versus 1.4 antihypertensive drugs; p=0.015). These results illustrate the challenges in ascertaining the BP-lowering efficacy of RDN when antihypertensive drugs are adjusted without a strict protocol.
Despite significant reductions in BP, half of the patients remained with uncontrolled hypertension and will require the addition of antihypertensive medications to attain guideline-recommended target values. There was large between-patient variability in the BP response, which might be due to variable renal nerve damage; however, there is currently no reliable perioperative marker of successful RDN.
The present analysis extends our knowledge about the efficacy of ultrasound RDN when carried out in patients with uncontrolled hypertension receiving first-line antihypertensive medication without a strictly prescribed protocol. Indeed, the majority of patients (76%) were treated with 1 or 2 antihypertensive drugs exclusively from common guideline-directed classes. These data are supported by the results of the SYPRAL HTN-ON MED trial11, in which the efficacy of radiofrequency RDN on BP was investigated in the presence of standard medical therapy (1, 2, or 3 antihypertensives), also prescribed at the discretion of the treating provider. The decrease in 24-hour ambulatory systolic BP at 6 months was significantly greater in the RDN group (–9.0 mmHg, 95% CI: –12.7 to –5.3 mmHg, p<0.0001) than in the sham group (–1.6 mmHg, 95% CI: –5.2 to 2.0 mmHg, p=0.365; baseline-adjusted difference between groups –7.0 mmHg, 95% CI: –12.0 to –2.1 mmHg, p=0.0059).
In the present analysis office BP reduction with RDN was more modest and characterised by pronounced variability (indicated by large standard deviations). However, ambulatory BP measurements are a better predictor of cardiovascular events than office BP measurements12 and have been recommended as the primary efficacy endpoints in device-based hypertension trials13,14,15. Particularly night-time BP, which may be caused by activation of the sympathetic nervous system, has been associated with worse cardiovascular outcomes12. A recent study suggested that higher night-time BP and a riser pattern of nocturnal BP were significantly associated with the risk of cardiovascular disease and especially heart failure16. The changes in night-time BP observed herein (2 months: –10.9±–14.1/–7.2±–9.4 mmHg; 6 months: –9.3±–15.6/–7.1±–10.6 mmHg) are therefore meaningful and may contribute to cardiovascular risk reduction in hypertensive individuals. Of note, reductions in ambulatory BP were greater when compared with office BP readings, which may be related to the fact that patients had to have a re-qualifying ambulatory but not office BP. In addition, office BP has greater variability and, given the small sample size in this study, ABP may better capture the true effect of RDN.
The incidence of adverse events in the crossover group was low. These data compare favourably with published information from randomised controlled trials and real-world studies17,18,19. The long-term follow-up of the multicentre, non-randomised, post-market study evaluating the safety and efficacy of ultrasound RDN (ACHIEVE) up to 12 months in patients with resistant hypertension documented sustained reductions in both office and ambulatory blood pressure and confirmed the safety of the procedure18. Also, the Global SYMPLICITY Registry, the largest available trial to date of outcomes after RDN in a hypertensive population, reported no long-term safety concerns following RDN17,20.
Limitations
Crossover patients and physicians were unblinded, so these data are subject to behavioural and/or medication-related effects that may have contributed to the observed results. Another limitation is that the majority of the patients enrolled in this study were Caucasian and applicability to other ethnicities may require further study. Adherence to medication is typically dynamic11. Objective assessment of adherence using chemical analysis was not part of the study protocol; however, this may better reflect real-world practice and outcomes.
FUTURE DIRECTIONS
The safety and efficacy of RDN beyond 6 months in patients who crossed over was not addressed in the present analysis; however, longer-term follow-up is ongoing and data from the cohort initially randomised to RDN showed durability of both the BP-lowering effect and safety of RDN up to 12 months. These long-term follow-up data from a randomised study representing real-world practice could be confirmed in larger registries.
Conclusions
Ambulatory daytime, night-time and 24-hour BP was significantly reduced in unblinded sham patients who subsequently crossed over and were treated with endovascular ultrasound RDN. While BP-lowering data with RDN are encouraging, for the majority of patients, RDN may need to be used in combination with antihypertensive medications in order to control BP adequately.
|
Impact on daily practice Patients included in the RADIANCE-HTN SOLO trial who crossed over and received ultrasound RDN had significant reductions in BP, consistent with the primary trial results. These data support the efficacy and safety of ultrasound RDN when performed under unblinded conditions in the presence of antihypertensive medication adjusted at the discretion of the investigator. |
Acknowledgements
The trial executive committee designed the protocol in conjunction with the sponsor. The sponsor was responsible for selection of clinical sites in collaboration with the executive committee, as well as collection, monitoring, and analysis of the data. The article was written by the lead author (Felix Mahfoud) with significant contributions from the co-authors. All authors had access to all the data, and Felix Mahfoud was responsible for the decision to submit the manuscript.
Funding
The study was funded by ReCor Medical, Inc. (Palo Alto, CA, USA).
Conflict of interest statement
F. Mahfoud is supported by Deutsche Gesellschaft für Kardiologie (DGK), and Deutsche Forschungsgemeinschaft (SFB TRR219) and has received scientific support and speaker honoraria from Bayer, Boehringer Ingelheim, Medtronic and ReCor Medical. M.J. Bloch has received personal fees from ReCor Medical and Medtronic. M. Azizi has received research grants from the French Ministry of Health, Quantum Genomics and the European H2020 program, has received grant support and non-financial support from ReCor Medical and Idorsia, and has received personal fees from CVRx. R.E. Schmieder has received grant support and personal fees from ReCor Medical, Medtronic, and Ablative Solutions. M.D. Lobo has received personal fees from ReCor Medical, Medtronic, CVRx, Ablative Solutions, Vascular Dynamics, ROX Medical and Tarilan Laser Technologies and grants from Medtronic. A.S.P. Sharp has received personal fees from ReCor Medical, Medtronic and Philips. J. Daemen has received grant support from ReCor Medical, Medtronic, Boston Scientific, Abbott Vascular, Acist Medical, AstraZeneca, Pie Medical, and PulseCath, and has received consulting fees from ReCor Medical, Medtronic, Acist Medical, Boston Scientific, Pie Medical, Siemens, and PulseCath. J. Basile has received grant support from ReCor Medical and Ablative Solutions. M.A. Weber has received personal fees from ReCor Medical, Medtronic, Boston Scientific, and Ablative Solutions. A. Scicli is an employee of ReCor Medical. C.K. McClure is an employee of NAMSA, a contractor for ReCor Medical. A.J. Kirtane reports institutional funding to Columbia University and/or Cardiovascular Research Foundation from Medtronic, Boston Scientific, Abbott Vascular, Abiomed, CSI, CathWorks, Siemens, Philips, and ReCor Medical. In addition to research grants, institutional funding includes fees paid to Columbia University and/or Cardiovascular Research Foundation for speaking engagements and/or consulting. He reports personal fees for consulting from Neurotronic, and travel expenses/meals from Medtronic, Boston Scientific, Abbott Vascular, Abiomed, CSI, CathWorks, Siemens, Philips, ReCor Medical, Chiesi, OpSens, Zoll, and Regeneron. Y. Wang has no conflicts of interests to declare.
Supplementary data
To read the full content of this article, please download the PDF.
