Abstract
Aims: Patients receiving long-term anticoagulant treatment with dabigatran may need to undergo a percutaneous coronary intervention (PCI). We studied markers of coagulation activation during elective PCI in patients using dabigatran in order to investigate whether coagulation activation upon balloon inflation and stenting is suppressed by dabigatran without additional heparin treatment.
Methods and results: This phase IIa, exploratory, multicentre, randomised, open-label study included 50 stable patients having an elective PCI. Patients on standard dual antiplatelet therapy (DAPT) were randomised (2:2:1) to either pre-procedural dabigatran 110 mg BID (n=19) or 150 mg BID (n=21), as compared to standard intraprocedural unfractionated heparin (UFH) (n=10). Following PCI, a significant increase in the levels of prothrombin fragment 1+2 (F1+2) in the combined dabigatran group was observed compared to the level just before the start of PCI (159.1 [1.4] pmol/l; geometric mean [gSD]). Levels at 0.5, 1.0, 1.5 and 2 hrs after the start of PCI ranged from 193.5 (1.4) to 270.6 pmol/l (1.7); (p-value for paired analysis=0.015, 0.022, 0.2342, 0.0379, respectively). Also, thrombin-antithrombin (TAT) complexes were increased significantly in the combined dabigatran group compared to pre-PCI levels (4.2 [2.2] ug/l). Levels ranged from 5.2 (2.5) to 8.5 (2.3) (p=0.0497, 0.0343, 0.005 and 0.1628, respectively). In contrast, in the control group of patients treated with UFH, no increase was observed in F1+2 and TAT complexes during PCI. Five out of 40 (12.5%) patients required bail-out anticoagulation in the dabigatran group, of whom four experienced a procedural myocardial infarction (MI), versus one out of 10 in the UFH group, who had a stent thrombosis without MI prior to the study-PCI. One minor access-site bleeding occurred in the dabigatran group.
Conclusions: Dabigatran treatment (110 mg or 150 mg BID) may not provide sufficient anticoagulation during PCI. ClinicalTrials.gov Identifier: NCT00818753 EudraCT. No: 2007-007536-25.
Introduction
Patient populations at risk of venous or arterial thrombosis, including patients with atrial fibrillation (AF), often receive long-term anticoagulant treatment. Many of these patients suffer co-existing atherosclerotic coronary artery disease (CAD) and may be in need of an urgent percutaneous coronary intervention (PCI) during the treatment period1-3.
During PCI more intensified anticoagulation may be needed to perform the procedure safely, because balloon inflation and stent placement can potentiate an existing prothrombotic state around lesion areas and lead to ischaemic complications4-7. The choice of the concomitant pharmacological environment is critical, as is the dosage of the drugs. The value of a periprocedural antithrombotic regimen depends on the balance between prevention of ischaemic and bleeding complications8.
Dabigatran etexilate, an orally available, potent, direct inhibitor of thrombin, given at a dose of 150 mg twice daily, is more effective than warfarin in the prevention of stroke and systemic embolism in patients with AF3. Dabigatran etexilate has little interaction with food and drugs and can therefore be prescribed in a fixed dose without the requirement of frequent monitoring9. It is as yet unknown whether patients who are on stable long-term anticoagulation with dabigatran are adequately anticoagulated to perform a PCI procedure safely or need additional (unfractionated) heparin to suppress coagulation activation during the procedure.
The primary goal of this study was to investigate whether dabigatran treatment, mimicked by giving a short course of 110 mg or 150 mg BID dabigatran before the procedure, prior to standard dual antiplatelet therapy (DAPT), adequately suppresses coagulation activation during elective PCI.
Methods
STUDY DESIGN AND POPULATION
The D-fine clinical trial is a phase IIa prospective, randomised, exploratory study conducted in four hospitals in The Netherlands. ClinicalTrials.gov Identifier: NCT00818753 EudraCT. Unique identifier No: 2007-007536-25. Patients were considered for the study if planned for a non-urgent PCI via femoral approach to treat symptomatic, obstructive CAD (demonstrated silent ischaemia, stable angina). The cardiac-specific troponin (cTn) at the time of the index PCI had to be below the 99th percentile of the upper reference limit (URL). Patients were excluded for lesion-specific conditions, if they were haemodynamically unstable or at increased bleeding risk (see Appendix for inclusion and exclusion criteria). Patients were followed for a maximum period of two weeks after the index PCI for this study.
An independent external safety physician (FV) was charged to monitor patient safety and advised the steering committee on potential issues that might have occurred during the conduct of the trial. Special emphasis was placed on capturing thrombotic intraprocedural complications such as abrupt vessel closure, no reflow, and formation of clots on or within the interventional equipment (i.e., catheters and wires). The trial was approved by the ethics committee at each participating institution, and all patients provided written informed consent.
RANDOMISATION AND STUDY TREATMENTS
Patients were randomly assigned through pre-prepared sealed envelopes, in an open-label fashion respecting a 2:2:1 ratio to either pre-procedural dabigatran (Boehringer Ingelheim Pharma GmbH & Co. KG, Ingelheim, Germany) 110 mg twice daily (n=19), dabigatran 150 mg twice daily (n=21) or intraprocedural unfractionated heparin (UFH) 70 IU per kilogram of body weight with subsequent boluses targeted to achieve an activated clotting time (ACT) of 250 to 300 (n=10). Dabigatran was started 24 hours before the index PCI for a total of three doses before PCI (respectively 24 hours, 12 hours and two hours before the index PCI). All patients were pre-treated with 75 to 325 mg aspirin, and clopidogrel (loading dose 300 mg) administered no less than 12 hours before the index PCI.
Given the overall low to medium-risk patient group included in this trial, glycoprotein IIb/IIIa inhibitor (abciximab: bolus of 0.25 mg per kilogram followed by an infusion of 0.125 µg per kilogram per minute; maximum dose, 10 µg per minute, for a maximum of 12 hours) use was restricted to a bail-out situation, as defined below, in addition to an additional bolus of heparin (50 U/kg maximum 3,500 U). Coronary stenting with either bare metal or drug-eluting stents, according to the choice of the physician, was the preferred method of PCI.
LABORATORY COAGULATION ASSAYS
The coagulation assay panel included the thrombin time performed with 10 units of thrombin (TT 10U, Thromboclotin; Siemens, Munich, Germany), prothrombin time (PT) and international normalised ratio (INR) (Tromborel S; Siemens), activated clotting time (ACT), activated partial thromboplastin time (aPTT, TriniCLOT; Trinity Biotech, Bray, Co. Wicklow, Ireland), fibrinogen according to Clauss (Thrombin Reagent; Siemens), D-dimer levels (AutoDimer; Biopool International, Ventura, CA, USA) and assays of thrombin formation: thrombin-antithrombin (TAT) complex (Enzygnost® TAT micro; Siemens), prothrombin fragment 1+2 (the activation peptide released from prothrombin during thrombin formation) (F1+2, Enzygnost® F1+2; Siemens). Dabigatran plasma levels were measured at screening (blank sample), just before second dose (12 hours before PCI), just before third dose (two hours before PCI), just before the index PCI (two hours post dose, around peak), six to eight hours post index PCI (eight to 10 hours post dose).
Blood samples for coagulation testing were collected in 3.2% trisodium citrate (0.105 M) using a vacutainer system (Becton Dickinson, Plymouth, UK). Blood samples for pharmacokinetic analysis were collected in K-EDTA (potassium ethylene-di-aminetetra-acetic acid). All samples were centrifuged within 30 minutes after collection at 4°C for 10 minutes at 2,000 g. Platelet-poor plasma was prepared frozen in small pre-labelled cryo vials and frozen immediately at –80°C until processing. Shipment of the frozen (–80°C) plasma samples from the clinical site to the central analytical laboratory was on dry ice. All coagulation testing was performed (in bath) at the Department of Haematology, Erasmus University Medical Center, Rotterdam, The Netherlands. The dabigatran plasma levels were measured by Boehringer Ingelheim, Biberach, Germany.
CLINICAL OUTCOME MEASURES
We assessed the number of patients who needed bail-out anticoagulant treatment for ischaemic cardiac events and/or had clinical or angiographic signs of catheter-related thrombosis during the PCI procedure such as abrupt vessel closure, new thrombus with reduced flow, no reflow; or clot formation on or within the interventional equipment (i.e., guiding catheter, guidewire).
We also assessed the occurrence of major and clinically relevant non-major in-hospital bleeding events or up to three days (whichever came first). Major bleeding events, clinically relevant non-major bleeding events, and minor bleeding events were classified on the basis of the Randomised Evaluation of Long-Term Anticoagulation Therapy (RE-LY)3 and the Thrombolysis in Myocardial Infarction (TIMI) scales10. The occurrence of other adverse events up to 14 days following the index PCI was captured. Myocardial infarction (MI) was defined according to the 2007 universal definition11.
The protocol issued for performance of a standard 12-lead electrocardiogram (before PCI, six to eight hours post PCI and 18-24 hours post PCI, at discharge and during the seven to 14-day follow-up visit), collection of blood samples for the measurement of cardiac enzyme levels (before PCI; six to eight hours post PCI and 18-24 hours post PCI) and the collection of haemoglobin levels.
STATISTICAL ANALYSIS
For the comparison of coagulation parameters over time, analysis of repeated measures was performed. The repeat measures model of the log-transformed laboratory values contained treatment, visit (time) and visit interaction as fixed classification effects, along with the log-transformed baseline laboratory value and log baseline lab value by visit interaction as linear covariates. An unstructured covariance structure was used to model the within-patient errors. Baseline to pre-procedure changes and pre-procedure to intervals during procedure changes, in log-transformed F1+2 and TAT laboratory values, were assessed.
For laboratory values, the repeated measures model reported results in adjusted geometric means and in the comparison of the ratio of these means between the treatments. The ratio was accompanied by a confidence interval and p-value. Due to log transformations, ratios were presented to achieve normality in the skewed distribution on the original scale.
Clinical outcome data were analysed according to a modified intention-to-treat principle, whereby patients who did not receive any study drug were excluded from all analyses, as was pre-specified in the protocol.
Differences between the treatments were expressed in odds ratios and their matching 95% confidence intervals. Interpolation of missing data was not conducted. Categorical outcomes were compared with the chi-square test or Fisher’s exact test. Continuous variables were compared using the Wilcoxon rank-sum test. ANOVA was used for between-group comparison of changes in F1+2 and TAT values; paired analysis of F1+2 and TAT lab values was assessed using the paired T-test.
Statistical analyses were performed using SAS software, version 9.2, by a dedicated statistician.
Results
PATIENT POPULATION AND TREATMENT ALLOCATION
A total of 53 patients were randomised between August 2009 and April 2010. Three patients in the dabigatran 110 mg dose group were excluded from the study: two had predefined major exclusion criteria (left main coronary artery disease, severe anaemia; both patients were excluded before the intake of any study medication) and one was not compliant to the intake of study medication. All other patients (n=50) completed the planned observation period.
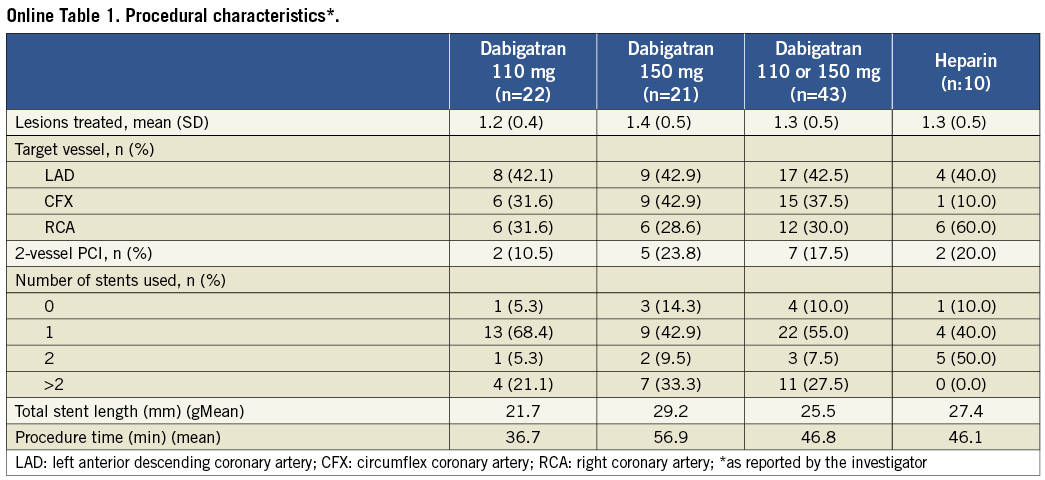
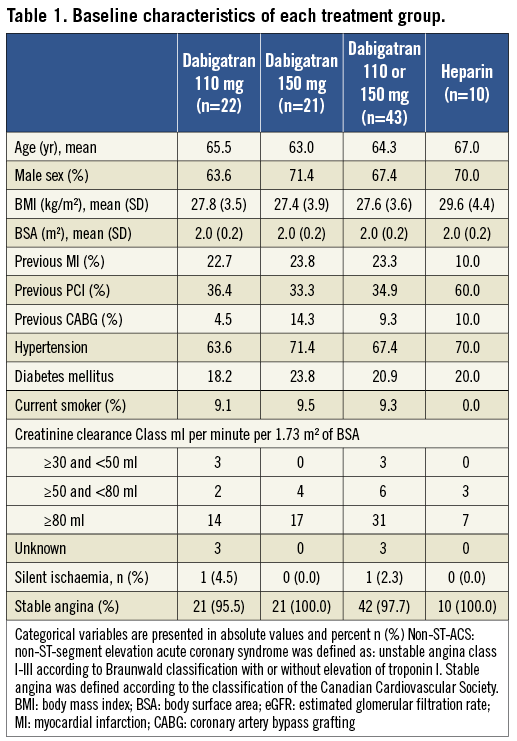
Baseline demographics are presented in Table 1. Median age was 65 (25th and 75th percentiles, 59 and 71 years), and 68% were male. The cardiac-specific troponin measurement at baseline was below the upper reference limit in all patients. The median estimated glomerular filtration fraction (eGFR) calculated by means of the Cockcroft-Gault formula was 95.4 ml/min (25th and 75th percentiles, 83.1 and 118.3 ml/min)12. The details of the procedural characteristics are listed in Online Table 1. All patients undergoing PCI (n=50) were pre-treated with DAPT. Of the 59 attempted lesions, all were successfully dilated. Three patients had a pressure-derived fractional flow reserve >0.80 and the lesions were left untouched. Drug-eluting stents were used in all except one patient (98%) and two-vessel stenting was performed in nine (18%) cases.
COAGULATION LABORATORY MEASUREMENTS
A selection of coagulation parameters is presented in Figure 1 and Table 2. The aPTT was significantly higher on heparin (p<0.0001 within the first two hours) as compared to the dabigatran groups. The APTT (seconds) increased from 28.8 (1.1) (geometric mean [gMean], standard deviation [gSD]) to a maximum of 51.8 (1.5 gSD) seconds at one hour following the start of PCI in the dabigatran group and an increase from 29.2 (1.1 gSD) to a maximum of 240 (1.0 gSD) seconds at one hour in the UFH group. A similar pattern was observed for ACT (Table 2). The INR was hardly influenced by dabigatran and remained below 1.5. The thrombin time (TT10U, seconds) at baseline was not different between the treatment groups. In the dabigatran group, the TT10U increased from 13.3 (1.1 gSD) to 60 (1.0 gSD) seconds after one hour. Also, in the heparin group, the TT10U increased from 14.6 (1.5 gSD) to 60 (1.0 gSD) seconds after an hour. During the interventional procedure a similar pattern was observed between dabigatran and UFH. However, after 18 hours, in the heparin group, the TT10U was normalised, whereas in the dabigatran group the TT10U was still slightly prolonged (23.8 [1.5 gSD] seconds).
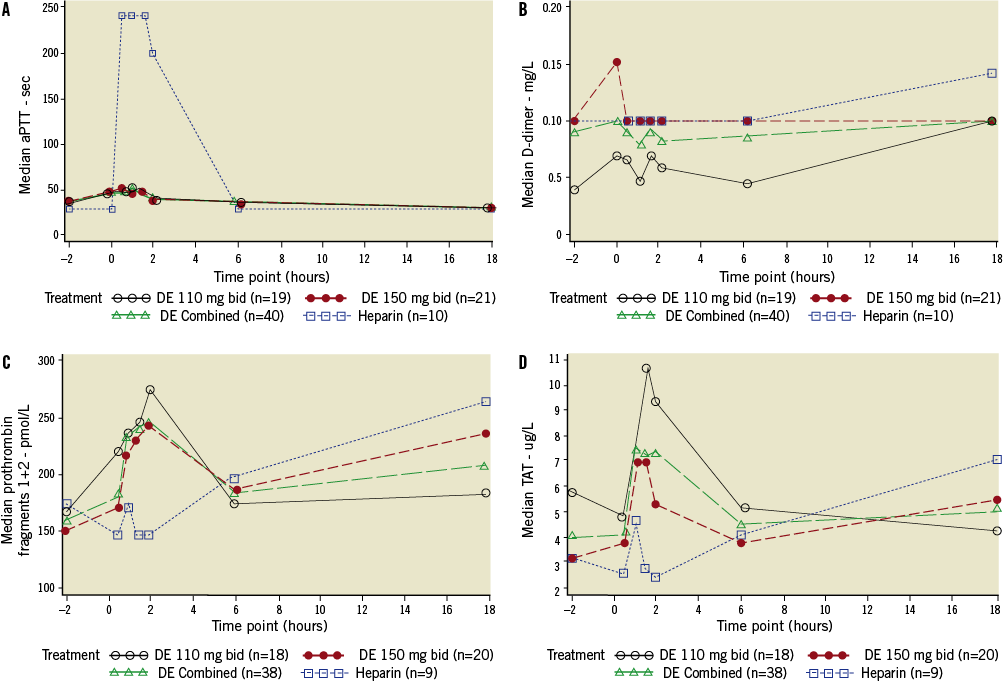
Figure 1. Results of markers of coagulation related to the index PCI. A-D: markers of coagulation activation for dabigatran vs. heparin. A) aPTT: activated partial thromboplastin time; B) D-dimers; C) F1+2: prothrombin fragments 1 and 2; D) TAT: thrombin-antithrombin III
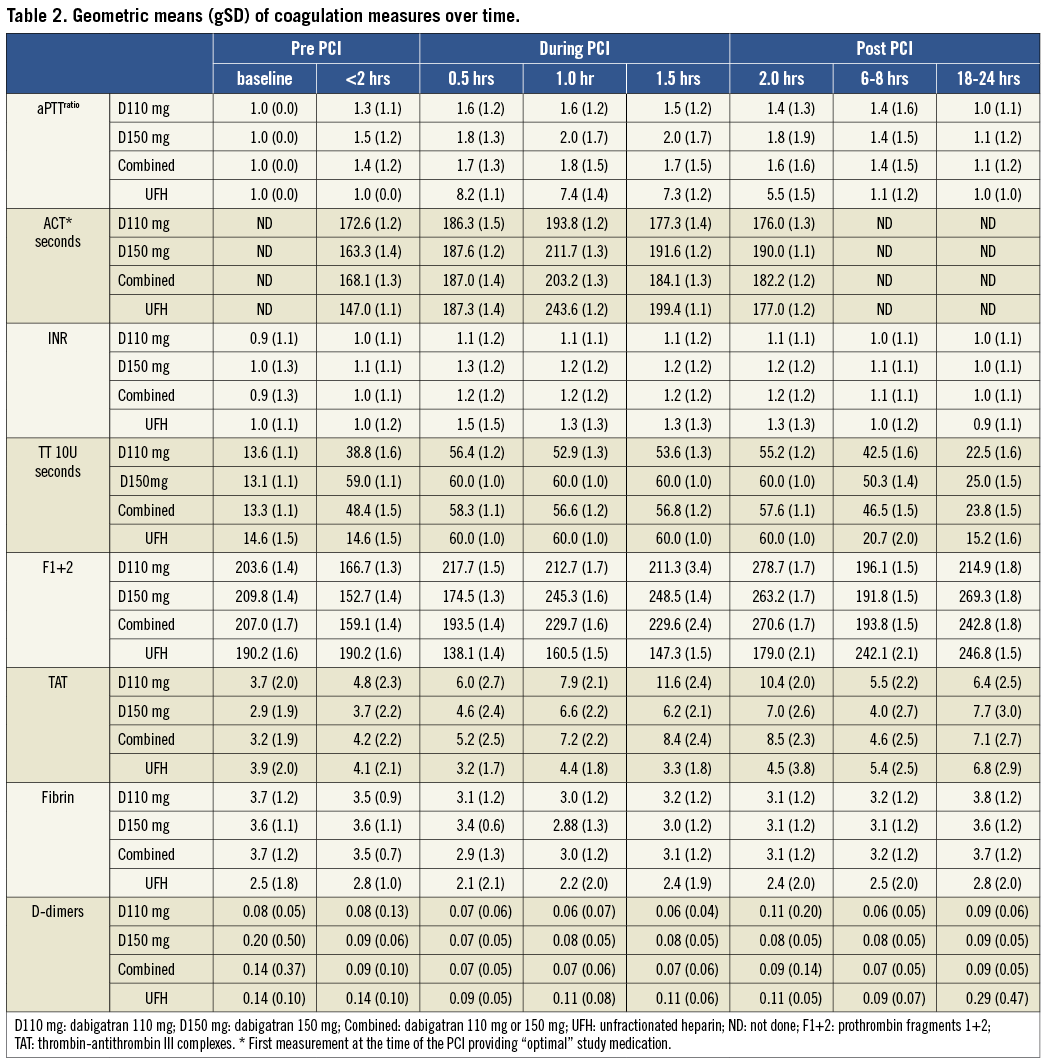
The relationship between dabigatran plasma concentrations and the coagulation parameters (INR, TT10U, aPTT and ACT,) is presented in Figure 2, panels A-D. This includes all blood samples taken during the study. We observed a weak correlation between dabigatran plasma levels and INR (r2=0.4029, p-value<0.0001) and ACT (r2=0.2981, p-value=0.0104). No correlation was found with TT10U (r2=0.0270, p-value=0.0818). The thrombin time could not be measured (>60 seconds, maximum) in most patients with high plasma concentrations of dabigatran (Figure 2B).
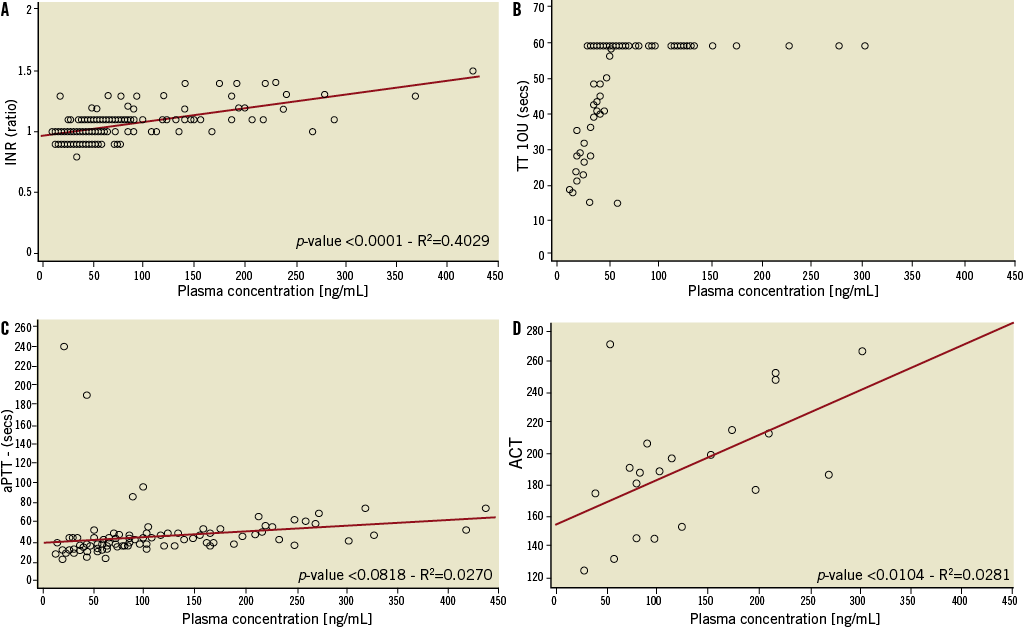
Figure 2. Relationship between dabigatran plasma concentrations and INR, TT 10U, aPTT and ACT. A-D: relationship between dabigatran plasma concentrations and the result of INR, TT10U, aPTT and ACT. A) INR: international normalised ratio; B) TT 10U: thrombin time (thrombin concentration 10 IU/ml); C) aPTT: activated partial thromboplastin time; D) ACT: activated clotting time
For the markers indicating coagulation activation, including F1+2 and TAT, there were no significant differences in pre-PCI values (Figure 1C-Figure 1D) (Table 2). However, a marked rise of both thrombin generation markers was observed in the dabigatran-treated patients as compared to the UFH group.
Following PCI, a significant increase in the levels of prothrombin fragment 1+2 (F1+2) in the combined dabigatran group was observed compared to the level at the start of PCI (159.1 [1.4] pmol/l; geometric mean [gSD]). Levels at 0.5, 1.0, 1.5 and 2 hours after the start of PCI ranged from 193.5 (1.4) to 270.6 pmol/l (1.7) (p-value for paired analysis =0.015, 0.022, 0.2342, 0.0379, respectively). Also thrombin-antithrombin (TAT) complexes were increased significantly in the combined dabigatran group compared to pre-PCI levels (4.2 [2.2] ug/l). Levels ranged from 5.2 (2.5) to 8.5 (2.3) (p=0.0497, 0.0343, 0.005 and 0.1628, respectively). In the control group of patients treated with UFH, no increase was observed in F1+2 and TAT complexes during PCI.
D-dimer levels were similar between the dabigatran and UFH groups and were not greatly increased during and after PCI.
PHARMACOKINETIC MEASUREMENTS
The dabigatran plasma concentrations within two hours prior to PCI (“trough”: 10-16 h post second dose and immediately before third dose) were 36.9 ng/ml (gMean) (45.1%gCV) for the 110 mg dose group and 64.2 ng/ml (44.0%gCV) for the 150 mg dose group. The levels remained within the same range at the time of PCI (“around peak”: one to three hours post third dose) (98.4 ng/ml, 89.9% gCV; 161 ng/ml, 65.2% gCV, respectively) and six to eight hours following PCI (36.6 ng/ml, 84.9% gCV; 63.1 ng/ml, 57.5% gCV, respectively). In patients with mild renal impairment (eGFR 30-80 ml/min) (5/43, 12%) dabigatran trough levels were approximately 42% higher (0.669 ng/mL/mg vs. 0.470 ng/mL/mg) and peak concentrations approximately 60% (1.86 ng/mL/mg vs. 1.16 ng/mL/mg) higher than in patients with an eGFR ≥80 ml/min.
CLINICAL OUTCOMES
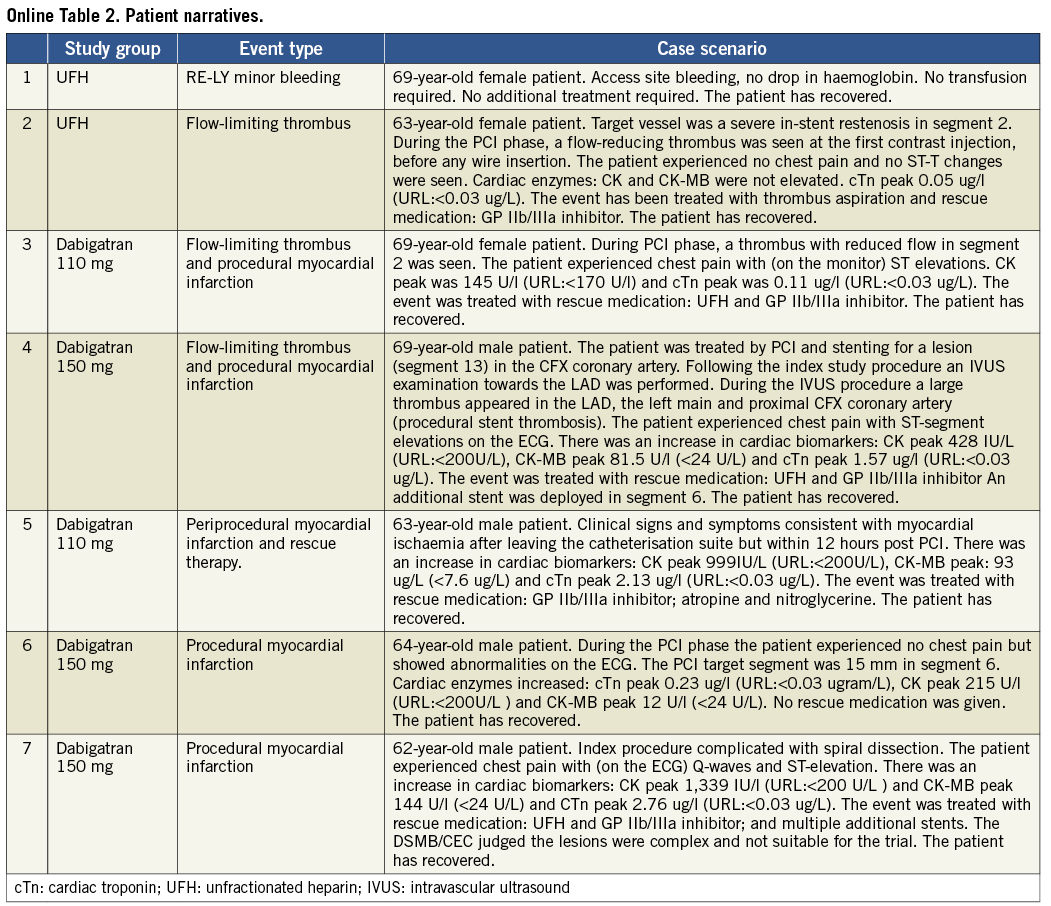
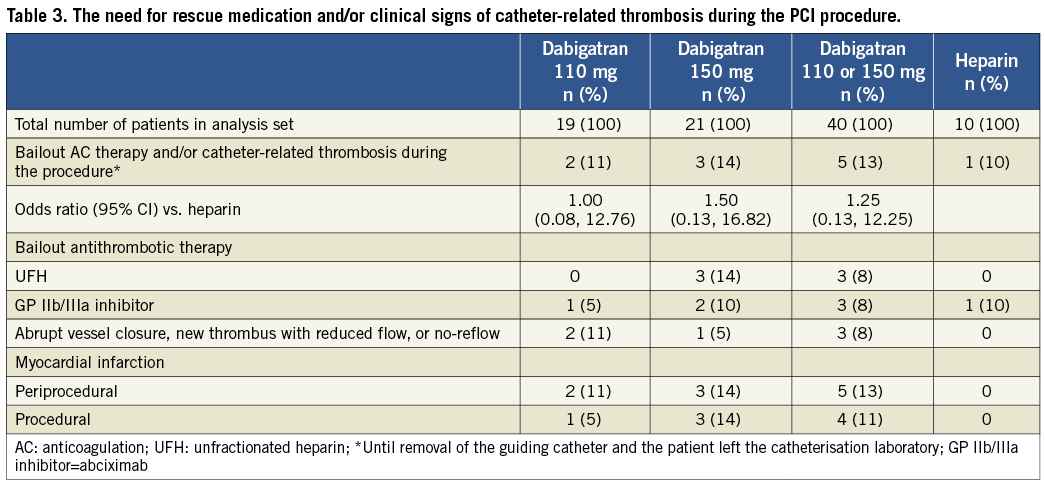
The overall proportion of patients who required rescue anticoagulant medication and/or had clinical signs of catheter-related thrombosis did not differ significantly between groups (odds ratio 1.25, 95% CI: 0.128-12.252) (Table 3). Two patients in the dabigatran group experienced a procedural flow-limiting thrombus formation imposing bail-out anticoagulation: one of these was a case of procedural stent thrombosis while an intravascular ultrasound examination was being performed. Additional (to the primary outcome) major adverse cardiac events up to 14 days occurred in two patients in the dabigatran groups (all were periprocedural myocardial infarction), versus none in the UFH group. One patient in the dabigatran 110 mg dose group developed new symptoms consistent with myocardial ischaemia after the patient left the catheterisation suite, but within 12 hours after the index PCI: a periprocedural MI was diagnosed. There were no deaths. For a full description of the individual patient narratives see Online Table 2. Major and clinically relevant bleeding events did not occur. The overall incidence of treatment-related (non-thrombosis or haemostasis) adverse events was similar in both treatment groups: 34.1% in the combined dabigatran group, 30.0% in the UFH group and of mild intensity. None led to discontinuation of the study drug (data not shown).
Discussion
The D-fine study is the first to investigate whether pre-procedural dabigatran provides sufficient anticoagulant effect to obviate the need for adjunctive heparin in case of PCI. Our results suggest that a short course of dabigatran, plus standard DAPT, but without concomitant intraprocedural UFH, prior to an elective PCI was insufficient to suppress coagulation activation during balloon inflation and stent expansion as compared to intraprocedural UFH.
Patients on oral anticoagulants in need of (an urgent) PCI represent a clinical conundrum, as management guidelines for revascularisation are based on clinical trials that have largely excluded patients who receive long-term anticoagulation therapy. Dabigatran etexilate is a new-generation oral direct thrombin inhibitor that decreases thromboembolic events in patients with atrial fibrillation3,13-14. The pharmacokinetic profile of dabigatran etexilate is characterised by maximum plasma concentrations at approximately two hours after oral administration, a bi-exponential distribution phase and a terminal half-life of about 11 hours in healthy volunteers above 65 years9. The total and peak exposure increase linearly and are dose-proportional after single and multiple oral dosing of the drug. Dose selection of dabigatran in this trial was based on phase II and III clinical trial data (i.e., RE-LY trial)15,16. In our study three doses of dabigatran (110 or 150 mg) were given before the PCI. This resulted in trough and peak dabigatran plasma concentrations that were 43% and 10% lower, respectively, than the steady state concentrations achieved in the previously reported RE-LY trial3. The trough concentrations were closer to those encountered in the VTE treatment study17. These differences in pharmacokinetics may be explained both by the short treatment course and by differences in study populations, mainly driven by differences in estimated glomerular filtration3,9.
In this mechanistic study the main focus was the periprocedural results of coagulation tests and levels of markers of coagulation activation in both the UFH and dabigatran-treated patients. The differential increase of levels of F1+2 and TAT complexes, which are both markers of thrombin generation, was significantly higher in patients treated with dabigatran than in those treated with UFH. This may have been caused by the fact that UFH inhibits the coagulation system not only via thrombin, but also via factor Xa and, to a lesser extent, also via other coagulation factors. Both dabigatran dosing groups had elevated TAT complex values as compared to UFH, suggesting more coagulation activation than in the heparin group. This seemed to be dabigatran dose-dependent, since patients treated with dabigatran 110 mg BID had F1+2 and TAT values that were even more increased than in those treated with dabigatran 150 mg BID (Figure 1).
Although our study was not powered to detect a difference in clinical outcome measures between UFH and dabigatran, the number of adverse events at 13% of patients is higher than that seen in general practice and in previous studies with other anticoagulants and may raise some concerns18,19. It is important to mention the recent report by Uchino et al who reported a 30% increased incidence of AMI in patients treated with dabigatran compared to vitamin K antagonists in a meta-analysis of all major dabigatran trials for various indications20.
The reported laboratory data, coupled with the cases of flow-limiting thrombus in the dabigatran groups, indicate that the differences between UFH and dabigatran with regard to the attenuation of coagulation activation may be clinically relevant. Whether higher doses of dabigatran etexilate, with or without an additional bolus of UFH, could support PCI remains a subject for further investigation. Pending further data, and analogous with the OASIS-5 trial and the HORIZONS-AMI trial, one may consider an additional bolus of heparin in patients in need of urgent PCI while being anticoagulated with dabigatran21,22.
Study limitations
This study has several limitations. First, the limitations of an open-label study design should be acknowledged: only the dabigatran dose was blinded. However, the laboratory technicians were not aware of the treatment the individual patients had received. This study was limited to elective PCI in stable patients. This consists of up to 60% of patients in routine PCI practice23. Patients with more complex lesion morphology and on-going myocardial ischaemia may be at higher risk of periprocedural thrombotic complications (complex lesion morphology, STEMI) and in need of even more intense periprocedural anticoagulation4,5. As indicated earlier, the D-fine study was only exploratory by design. D-fine was not powered to show a difference in clinical or angiographic thrombotic complications among its three arms. Overall, the trial is too small to draw any conclusion regarding differences in clinical outcome. A definitive non-inferiority study versus UFH in an all-comers PCI setting will require a considerable number of patients and may not be achievable.
In conclusion, in the setting of PCI, a short course of dabigatran prior to the procedure, without intraprocedural heparin, may not be sufficient to suppress coagulation activation compared to intraprocedural unfractionated heparin.
Acknowledgements
The authors recognise the valuable input of K. Metzman, PhD (pharmacokinetic data analysis and interpretation), L.M.C. van Campen, MD (source verification of the data) and P. Gobbels (statistical data analysis). We thank G.A. Van Es for his critical revision of the intellectual content of the manuscript. We thank the study staff at the participating centres for their efforts in collecting the data and the patients who volunteered to participate in the study.
Funding
The members of the steering committee, including the representatives of the sponsor, were responsible for the design and conduct of the study and the writing of the paper. The sponsor was responsible for collection and source verification of the data, with oversight by an independent clinical events committee. Analyses were performed by the sponsor and were verified by an independent statistician, Wietze Lindeboom.
Conflict of interest statement
J. Friedman, P. Reilly, and R. L. Jones are employees of Boehringer Ingelheim. The other authors have no conflicts of interest to declare.
Online data supplement
Appendix. Patients included in this study.
Table 1. Procedural characteristics.
Table 2. Clinical outcomes, patient narratives.
Appendix
PATIENT SELECTION
INCLUSION CRITERIA:
Patients were eligible for the D-fine study if they were:
1. between 18 and 85 years of age inclusive
2. due to undergo an elective (non-urgent) PCI on one or multiple lesions in the native coronary vessel(s) via a femoral approach
3. if they provided informed consent.
EXCLUSION CRITERIA:
Patients were excluded from the D-fine study if they:
1. were not a candidate for PCI or due to lesion-specific conditions:
a. left main disease
b. chronic total occlusions
c. bifurcation lesions, with haemodynamic important side branch*
d. three-vessel disease requiring treatment of more than two lesions (no staging is allowed)
2. had to undergo PCI for restenosis**
3. had haemodynamic instability
4. had severe hypertension not adequately controlled by antihypertensive therapy at the screening visit (blood pressure >180/110 mmHg)
5. had significant mitral or aortic valve disease
6. had increased bleeding risk:
a. ischaemic stroke within the last year or history of any previous haemorrhagic stroke, or
b. intracranial aneurysm
c. recent (<1 month) trauma or major surgery (including bypass surgery) in the last three months
d. symptomatic or endoscopically documented ulcer disease in the previous 30 days
e. active or recent (<3 months) major, minor TIMI-definition bleeding
f. impaired haemostasis: thrombocytopenia (platelet count <100*109) or active bleeding disorder
g. anticoagulant (heparin, coumarin) use; international normalised ratio >1.5
h. use of oral non-steroidal inflammatory drugs (NSAID) within 12 hours prior to PCI
i. past or present bleeding disorder (including congenital bleeding disorders such as von Willebrand’s disease or haemophilia; acquired bleeding disorders; and unexplained clinically significant bleeding disorders)
j. thrombolytic therapy within 24 hours preceding randomisation
7. had severe renal impairment (e.g., known creatinine clearance <30 mL/min (GFR - assessed by the Modification of Diet in Renal Disease [MDRD] study equation) or clinical markers of severe renal impairment) [R02-2429] [R02-2529]
8. had known liver disease (e.g., known high concentrations of liver enzymes [more than twice the ULN])
9. were pre-menopausal (last menstruation <1 year prior to screening) sexually active women who: were pregnant or nursing or were not surgically sterile or were of child-bearing potential and not practicing an acceptable method of birth control (acceptable methods included intrauterine devices [IUD], oral, implantable or injectable contraceptives, double barrier or vasectomised partner). A pregnancy test indicated pregnancy in a woman of child-bearing potential at screening.
10. had been treated with other investigational drugs or devices within 30 days before enrolment or
11. had planned use of investigational drugs or devices during the study.
12. were using quinidine
13. had a known allergy, hypersensitivity or contraindication to clopidogrel, aspirin
14. were unable to give informed consent or had a high likelihood of being unavailable for follow-up:-
a. inability or unwillingness to perform 7 to 14-day follow-up
b. patients considered unreliable by the investigator concerning the requirements for follow-up during the study and/or compliance with study drug administration or had any condition which, in the opinion of the investigator, would not allow safe participation in the study (e.g., drug addiction, alcohol abuse).
PROTOCOL AMENDMENT 17/11/2009:
* Simple bifurcation lesions (Medina class: 1,0,0, 0,1,0, 1,0,1 and angle between the distal main vessel and the side branch >70°), with a minimal diameter of the side branch of >2.0 mm by visual estimate. (In all cases of side branch stenting the operator is required to attempt a “kissing balloon” dilatation at the end of the procedure).
** PCI for diffuse restenosis (focal in-stent restenosis can be treated in this study)