
Abstract
Aims: The aim of this study was to evaluate the efficacy and safety of the BioNIR stent compared with the Resolute Integrity stent for the treatment of coronary artery disease.
Methods and results: This first-in-human, multicentre, single-blind randomised non-inferiority trial was performed in Europe and Israel. Patients with stable coronary artery disease or acute coronary syndromes were randomly assigned to treatment with BioNIR or Resolute Integrity stents in a 2:1 fashion. The primary endpoint was angiographic in-stent late lumen loss (LLL) at six months. Three hundred and two patients were randomised, of whom 261 (86.0%) underwent six-month angiographic follow-up. The BioNIR stent was non-inferior to the Resolute Integrity stent for the primary endpoint of in-stent LLL at six months (0.04±0.30 mm vs. 0.03±0.31 mm, respectively, pnoninferiority<0.0001). At 12-month follow-up, target lesion failure occurred in 3.4% in the BioNIR group and 5.9% in the Resolute Integrity group (p=0.22). Rates of MACE were similar between the BioNIR and Resolute Integrity groups (4.3% vs. 5.9%, respectively, p=0.45).
Conclusions: The BioNIR stent was non-inferior to the Resolute Integrity stent for the primary endpoint of angiographic in-stent LLL at six months. Clinical outcomes at one year were comparable between the two groups. Clinical Trials Identifier: NCT01995500
Abbreviations
DAPT: dual antiplatelet therapy
DES: drug-eluting stent
LLL: late lumen loss
MACE: major adverse cardiac events
MI: myocardial infarction
MLD: minimal lumen diameter
PCI: percutaneous coronary intervention
RES: ridaforolimus-eluting stent
TLF: target lesion failure
TVR: target vessel revascularisation
ZES: zotarolimus-eluting stent
Introduction
Early-generation drug-eluting stents (DES) improved the clinical outcomes of patients undergoing percutaneous coronary intervention (PCI) and reduced neointimal hyperplasia and restenosis compared to bare metal stents1-3. However, this antirestenotic effect came at the expense of delayed arterial healing of the vessel and an increased risk of late adverse events. New-generation DES overcame the limits of earlier devices by using more biocompatible polymer coatings, thinner struts and different antiproliferative agents and dosages4. The NIREUS trial investigated the BioNIR stent (Medinol, Tel Aviv, Israel), a new drug-eluting stent eluting the rapamycin analogue ridaforolimus. The study aimed to assess whether the BioNIR stent is non-inferior compared to the Resolute Integrity zotarolimus-eluting stent (Medtronic Cardiovascular, Santa Rosa, CA, USA) in terms of six-month angiographic in-stent LLL and clinical events. This study serves as the first-in-man experience with this stent.
Methods
TRIAL AND STUDY POPULATION
The NIREUS study (BioNIR ridaforolimus-eluting coronary stent system EUropean angiography Study; clinicaltrials.gov identifier NCT01995500) was a prospective, single-blind, non-inferiority multicentre trial that randomised patients in a 2:1 ratio to receive either the BioNIR ridaforolimus-eluting stent (RES) or the Resolute Integrity zotarolimus-eluting stent (ZES). The trial was conducted at 31 hospitals in Europe and in Israel. Major angiographic inclusion criteria were a reference vessel diameter between 2.5 and 4.25 mm and a lesion length less than 28 mm. Treatment of a maximum of three de novo lesions per patient was permitted. Important exclusion criteria included ST-segment elevation myocardial infarction, a left ventricular ejection fraction <30%, prior target vessel PCI within 12 months of the index procedure, treatment of bypass grafts and additional complex PCI conditions. Institutional review boards at each enrolling site approved the study and written informed consent was obtained prior to PCI.
DEVICE DESCRIPTION
The BioNIR stent consists of a cobalt-chromium alloy platform with struts of variable width (the wide cells measure 72 µm and the narrow cells 40 µm) (Figure 1); strut thickness is 87 µm. The wide cells were designed to provide radial strength whereas the narrow cells provide flexibility. The variable strut widths enable differential lengthening of stent cells to accommodate uniform drug distribution in variable vessel anatomies, and they enhance conformability.

Figure 1. BioNIR stent geometry. The BioNIR stent is constructed of cobalt-chromium struts that are 87 µm thick. The struts are of variable width enabling differential lengthening. A) Longitudinal view of a BioNIR stent showing narrow and wide struts. B) Cross-section through a stent crimped on a balloon showing narrow struts (42 µm). C) Cross-section through a stent crimped on a balloon showing wide struts (72 µm).
The stent is coated with a proprietary durable polymer matrix, composed of Poly (n-butyl methacrylate) (PBMA) and CarboSil® (DSM Biomedical, Exton, PA, USA). The coating has elastomeric properties that enable elastic expansion and contraction without cracking or tearing. The elastomeric polymer allows uniformity of antiproliferative drug distribution and elution5. The coating contains ridaforolimus at a concentration of 1.1 µg/mm², with near complete drug elution within 90 days.
RANDOMISATION, INTERVENTIONAL PROCEDURE, ADJUNCTIVE DRUG THERAPIES AND ANGIOGRAPHIC FOLLOW-UP
Patients were blinded to treatment assignment and randomised to the RES or ZES groups in a 2:1 fashion. Randomisation was stratified according to the presence of medically treated diabetes and study site.
A 6 Fr or larger guide catheter had to be used for accurate qualitative and quantitative coronary analysis (QCA) measurements6. Following intracoronary injection of nitroglycerine (50-200 mcg was recommended), baseline angiography of the involved vessel(s) was performed for at least two orthogonal views showing the target lesion free of foreshortening or vessel overlap according to the angiographic core laboratory guidelines.
Before revascularisation, all patients received treatment with aspirin and clopidogrel, ticagrelor or prasugrel, according to the operator’s discretion. Six months of dual antiplatelet therapy (DAPT) was mandatory. Anticoagulation and glycoprotein IIb/IIIa inhibitor therapies were left to the operator’s discretion. In-hospital, 30-day, and 12-month clinical follow-up, and six-month angiographic follow-up were scheduled after the index procedure.
DATA MANAGEMENT AND CORE LABORATORIES
Data were submitted to a central data coordinating facility (Cardiovascular Research Foundation, New York, NY, USA). Baseline and follow-up coronary angiograms were reviewed by an independent core laboratory (Cardiovascular Research Foundation). The angiographic core lab performed QCA assessment of all target lesions pre procedure, post procedure and at six-month follow-up (CAAS Workstation, v. 5.11.2 software; Pie Medical Imaging, Maastricht, the Netherlands). In-segment and in-stent target lesion minimal lumen diameter (MLD) and diameter stenosis (DS) were obtained by QCA. LLL was calculated for matched baseline and follow-up angiograms. After receiving the results of LLL that were calculated with the CAAS software, and in view of the lower than expected LLL results for both stents, we performed a second QCA analysis of LLL using QAngio XA, Version 7.2.34.0 software (Medis medical imaging systems bv, Leiden, the Netherlands). The methods used to calculate LLL with the second software were identical.
STUDY ENDPOINTS AND DEFINITIONS
The primary endpoint in-stent LLL was defined as the difference (in mm) between the post-procedure and the six-month follow-up angiography MLDs as measured by the angiographic core laboratory.
Secondary angiographic efficacy endpoints at six-month follow-up included in-segment LLL, percent DS and angiographic binary restenosis (both in-stent and in-segment). Binary restenosis was defined as a stenosis ≥50% of the lumen diameter of the target lesion. Restenosis patterns were characterised according to Mehran’s classification7.
Secondary clinical endpoints included major adverse cardiac events (MACE; cardiac death, any myocardial infarction [MI] and clinically driven target lesion revascularisation [TLR]), target vessel failure (TVF; all-cause death, target vessel-related MI and clinically driven target vessel revascularisation [TVR]), and target lesion failure (TLF; cardiac death, target vessel-related MI and clinically driven TLR). Additional secondary endpoints included the individual components of the composite endpoints, and stent thrombosis according to Academic Research Consortium criteria8.
Periprocedural MI was defined according to Society of Coronary Angiography and Interventions criteria9, and spontaneous MI was defined according to the universal definition of myocardial infarction10.
Clinically driven revascularisation was identified as any repeat revascularisation of the target lesion or target vessel associated with either: 1) ischaemic symptoms and/or an abnormal functional study and a ≥50% coronary stenosis by quantitative angiography; or 2) any revascularisation of a ≥70% coronary stenosis by quantitative angiography. An independent clinical events committee adjudicated all primary and secondary clinical endpoints.
STATISTICAL ANALYSIS
This randomised study was designed to determine the non-inferiority of RES compared with ZES with respect to the primary endpoint of six-month in-stent LLL. Based on previous data11-13 and assuming a linear accumulation between eight and 13 months, the anticipated LLL value for both stent cohorts was 0.20±0.44 mm, and a non-inferiority margin of 0.20 mm was set. With the assumption of 25% loss to follow-up, a sample size of 300 patients was required for the trial to have 89% statistical power to demonstrate non-inferiority at a one-sided alpha level of 0.025. Patients were analysed on the basis of the intention-to-treat principle. Baseline characteristics were summarised in terms of frequencies and percentages for categorical variables and by means with standard deviations for continuous variables. Categorical variables were compared by Fisher’s exact test. Continuous variables were compared by the two-sample t-test. Cumulative event-free survival was summarised as Kaplan-Meier estimates. A p-value of 0.05 was considered statistically significant. All analyses were performed with SAS software, version 9.4 (SAS Institute, Cary, NC, USA).
Results
PATIENT ENROLMENT AND BASELINE CHARACTERISTICS
Three hundred and two patients were randomised in a 2:1 fashion, of whom 261 patients (86%) had a qualifying angiogram performed at six months.
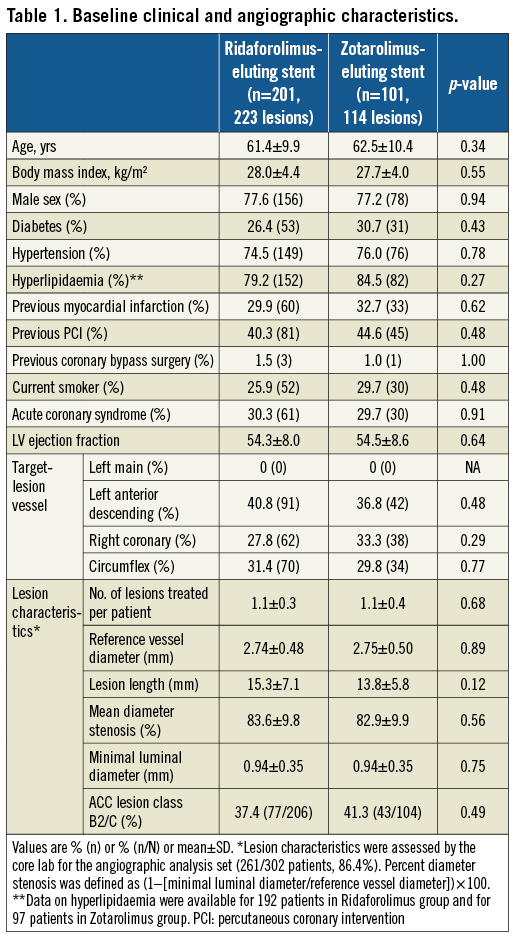
The baseline clinical and angiographic characteristics of the patients were well balanced between the two groups. Approximately one third had diabetes and almost one third presented with an acute coronary syndrome (Table 1).
PROCEDURAL OUTCOMES
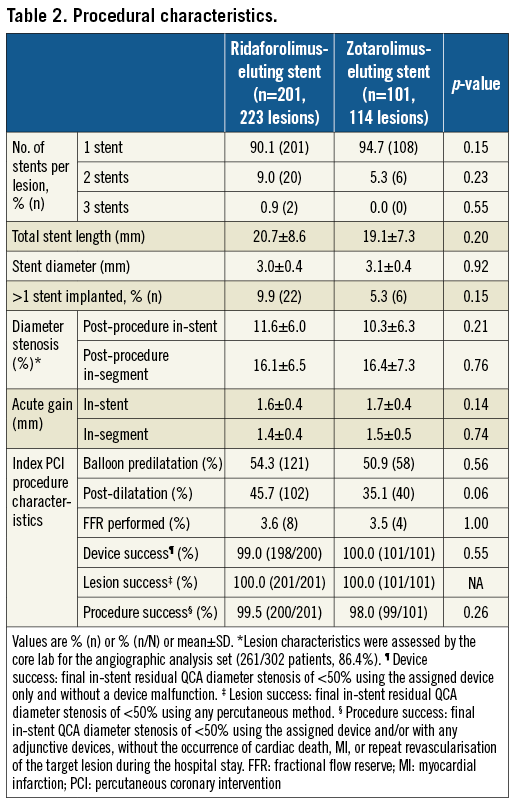
As shown in Table 2, the number of stents per patient, stent length and stent diameter were similar in both treatment groups. Overall procedural (99.0%) and device success (99.3%) were high and similar for both stent types (device success was not achieved in two patients of the BioNIR group due to failure to cross the lesion; procedural success was not achieved in one patient of the BioNIR group and two patients of the Resolute Integrity group due to periprocedural MI).
ANGIOGRAPHIC RESULTS
The primary endpoint, in-stent late lumen loss at six months, was 0.04±0.31 mm and 0.03±0.31 mm in the RES and ZES groups, respectively (p=0.79) (Figure 2), thus achieving the predefined non-inferiority margin (pnoninferiority<0.0001).
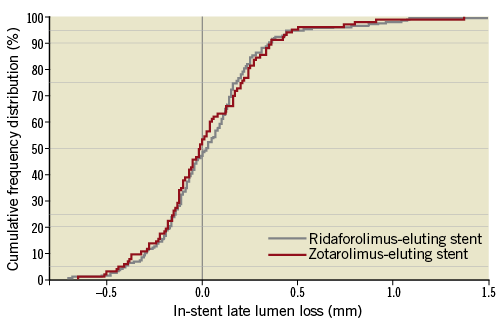
Figure 2. In-stent late lumen loss at six months. Cumulative frequency distributions of in-stent late lumen loss by stent type: ridaforolimus-eluting stent (grey line) and zotarolimus-eluting stent (red line).
Secondary angiographic endpoints both in-stent and in-segment were comparable between the RES- and ZES-eluting stent groups (Table 3). Overall in-segment binary restenosis rates were 3.4% in the RES group and 3.8% in the ZES group (p=0.84).
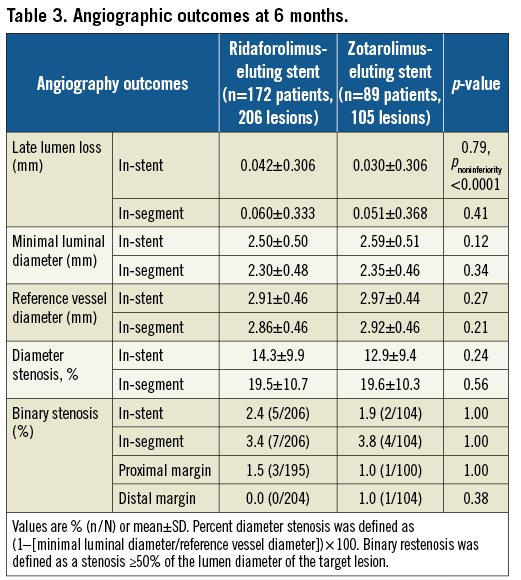
Restenosis patterns were similar and usually focal, with no observed cases of total occlusions and one case of diffuse restenosis in each group (0.6% of lesions).
Among the pre-specified subgroup of diabetic patients, no significant differences were observed in the rates of LLL at six months, that were 0.17±0.34 mm and 0.05±0.37 mm in the RES and ZES groups, respectively (p=0.28).
THIRTY-DAY AND ONE-YEAR CLINICAL OUTCOMES
Clinical results are presented in Table 4. At 30 days, there were no deaths in either group, and rates of MACE, TVF and TLF were low and similar in both groups.
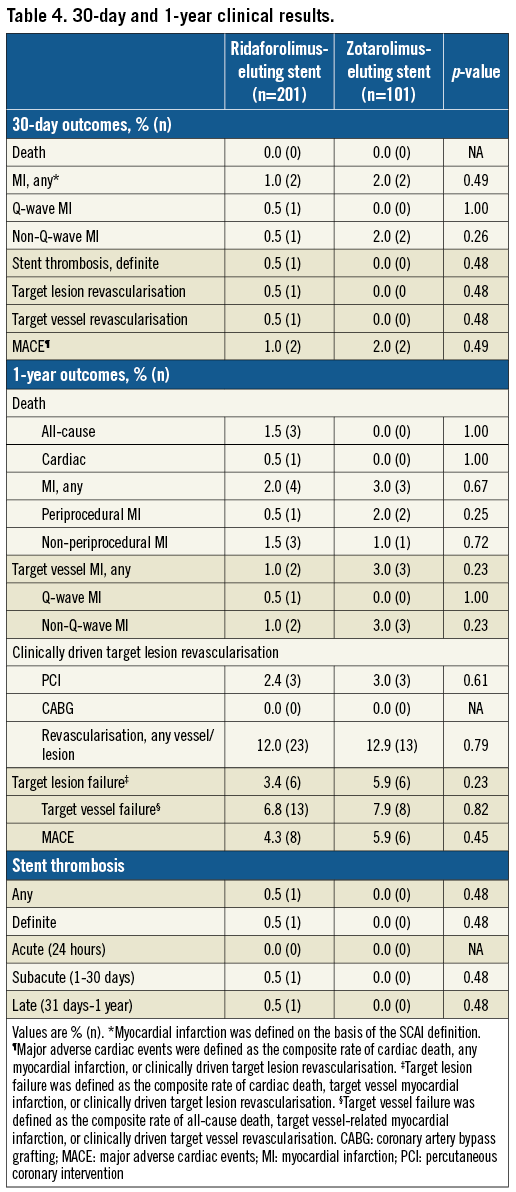
No additional differences in clinical outcomes were found at 12 months between the two groups. The composite endpoint of TLF at 12 months was 3.4% in the RES group and 5.9% in the ZES group (p=0.22). Rates of MACE and TVF were similar in both groups.
At 12 months, revascularisation occurred in 12.0% of the RES group and 12.9% of the ZES group (p=0.91). Of these events, clinically driven TLR procedures occurred in 2.4% and 3.0% of RES and ZES patients, respectively (p=0.68). Overall, 61% of the PCIs were in non-target vessels.
Definite stent thrombosis occurred in a single patient treated with RES in the NIREUS trial. Non-compliance with DAPT was confirmed in this case.
Discussion
NIREUS is the first randomised trial directly comparing the BioNIR RES with an established durable polymer DES, the Resolute Integrity ZES. The main findings of our study are: 1) in-stent LLL for the BioNIR stent was 0.04±0.30 mm and was 0.03±0.31 mm for the Resolute stent, pnoninferiority<0.0001; 2) similar safety and efficacy profiles were found between the two treatment groups and low clinical event rates were documented at 12 months. The primary endpoint of the study was in-stent LLL at six months after implantation. LLL is considered an established predictor of the long-term clinical efficacy of DES and a strong predictor of binary restenosis and TLR14. As a consequence, given the low rates of TLR accomplished by most current DES, which require large patient populations to prove the clinical non-inferiority or superiority of a new device, the use of angiographic parameters as a surrogate appears reliable. The low rates of LLL shown by the new BioNIR RES are reassuring and were confirmed by the recently completed BIONICS trial, a 1,919-patient trial designed to investigate clinical outcomes with RES compared with the Resolute ZES. The trial, including patients with more complex, “off-label” clinical indications and lesion anatomy, showed that the novel RES was non-inferior to ZES in terms of TLF with similar measures of LLL at one-year follow-up15.
In-stent LLL was found to be remarkably low for both stents with a mean of 0.03-0.04 mm. The results presented here are favourable compared with previously reported studies evaluating contemporary DES using limus drugs16-19. Moreover, in NIREUS, the extensively investigated Resolute Integrity ZES showed significantly lower LLL at six months when compared to previous trials of this stent (0.22 mm and 0.29 mm)20,21. Differences in core laboratories in terms of methodology and software used might have played a role in the overall excellent angiographic findings of both devices. The timing of LLL measurement (six months) and the patient characteristics of the NIREUS trial were not identical to those of other trials reported, and this may also have contributed to the lower rates of in-stent LLL. Collectively, these results suggest that the efficacy of the BioNIR stent is acceptable. This is also reflected in the low rates of clinical events up to 12 months.
The acute procedural success was 99.5% in the RES group vs. 98% in the ZES group (p=0.26), suggesting similar deliverability of both devices. With regard to safety, RES and ZES were associated with comparable risks of adverse events. The overall 12-month MACE rate was 4.3% vs. 5.9% (p=0.45) for the RES and ZES, respectively. The NIREUS trial was not powered to detect differences in clinical outcomes; we did not observe any statistical differences in clinical events. Nonetheless, rates of TVF and TLF with the BioNIR stents compared favourably to the rates with ZES supporting the angiographic findings. The 3.4% TLF rate with the RES at one year compares well to previous studies with durable polymer stents5,22.
Different stent designs, polymer coatings and antiproliferative agents may translate into differences in device performance and clinical outcomes. The BioNIR’s unique stent design capable of uniform scaffolding and drug release may have played an important role in reducing delayed healing and inflammatory reactions, resulting in the low rates of LLL and TLF observed at one year. Rates of stent thrombosis were low, with a single case occurring in the ridaforolimus group. The patient, re-admitted at nine days post stent implantation, reported non-compliance to DAPT, which has been associated with a fivefold increase in rates of stent thrombosis11.
Overall, the NIREUS results were obtained within a broad patient population, providing evidence for the safety and efficacy of the BioNIR stent in a wide variety of patients.
Study limitations
The NIREUS trial was powered for an angiographic endpoint and does not allow drawing firm conclusions concerning clinical outcomes. Second, despite the broader enrolment criteria adopted compared to other registration studies, STEMI patients within the first 24 hours and more complex lesions were excluded. Finally, longer follow-up than one year is required to prove long-term efficacy of the BioNIR stent.
Conclusions
This report on the use of the BioNIR RES provides evidence for the safety and efficacy of the study device in a relevant broad population. The NIREUS trial demonstrated good performance, including strong suppression of neointimal proliferation at six months and high post-procedural success of the BioNIR RES. These promising results were further confirmed by the clinical outcomes reported by the larger BIONICS trial.
| Impact on daily practice The NIREUS trial provides evidence of good suppression of in-stent late lumen loss at six months with the use of a novel ridaforolimus-eluting stent, which was non-inferior to that achieved with a zotarolimus-eluting stent. |
Funding
This trial was funded by Medinol Ltd., Tel Aviv, Israel.
Conflict of interest statement
O. Ben-Yehuda is an employee of the Cardiovascular Research Foundation (CRF) which received funding from Medinol Ltd for the conduct of the trial. M. Jonas is a consultant for Medinol. G. Perlman is an employee of Medinol. D. Kandzari is a consultant for Medtronic, Boston Scientific, Micell and St. Jude Medical, and has received scientific research support from Medtronic, Abbott Vascular, Boston Scientific, St. Jude Medical, Medinol, and Biotronik. P. Smits is a consultant for Medinol, Abbott Vascular, and St. Jude Medical, and has received research support from Abbott Vascular, St. Jude Medical, and Terumo. The other authors have no conflicts of interest to declare.
