
Abstract
Aims: Bindarit (BND) is a selective inhibitor of monocyte chemotactic protein-1 (MCP-1/CCL2), which plays an important role in generating intimal hyperplasia. Our aim was to explore the efficacy and safety of bindarit in preventing restenosis following percutaneous coronary intervention.
Methods and results: A phase II, double-blind, multicentre randomised trial included 148 patients randomised into three arms (BND 600 mg, n=48; BND 1,200 mg, n=49; PLB, n=51). Bindarit was given following PCI and continued for 180 days. Monthly clinical follow-up and six-month coronary angiography were conducted. The primary endpoint was in-segment late loss; the main secondary endpoints were in-stent late loss and major adverse cardiovascular events. Efficacy analysis was carried out on two populations, ITT and PP. There were no significant differences in the baseline characteristics among the three treatment groups. In-segment and in-stent late loss at six months in BND 600, BND 1,200 and PLB were: (ITT 0.54 vs. 0.52 vs. 0.72; p=0.21), (PP 0.46 vs. 0.53 vs. 0.72; p=0.12) and (ITT 0.74 vs. 0.74 vs. 1.05; p=0.01), (PP 0.66 vs. 0.73 vs. 1.06; p=0.003), respectively. The MACE rates at nine months among treatment groups were 20.8% vs. 28.6% vs. 25.5% (p=0.54), respectively.
Conclusions: This was a negative study with the primary endpoint not being met. However, significant reduction in the in-stent late loss suggests that bindarit probably exerts a favourable action on the vessel wall following angioplasty. Bindarit was well tolerated with a compliance rate of over 90%. A larger study utilising a loading dose and targeting a specific patient cohort may demonstrate more significant results.
Introduction
One of the major constraints of coronary stents is their vulnerability to restenosis due to intimal hyperplasia1,2. Although in-stent restenosis (ISR) has been significantly reduced owing to improvements in stent technology, including the introduction of drug-eluting stents3-5, it cannot be completely eliminated since the trigger for intimal hyperplasia begins with balloon injury to the vessel wall and is aggravated by the presence of metal struts6-8. In addition, there are certain clinical settings where the use of drug-eluting stents is considered not ideal or has limited applicability, such as in peripheral interventions9-11. MCP-1/CCL2 has been found to play an important role in the genesis of intimal hyperplasia12-14. It exhibits potent chemotactic activity towards circulating monocytes and T-lymphocytes and directs them to areas of vessel injury to promote smooth muscle cell proliferation. Increased levels of circulating MCP-1/CCL2 have been demonstrated in patients with restenosis as compared to those without restenosis15-17. Inhibition of MCP-1/CCL2 synthesis either by gene deletion or by blocking the MCP-1/CCL2 receptor in animal models has been shown to attenuate smooth muscle cell proliferation15-18. This notion of targeting MCP-1/CCL2 might be the key to preventing restenosis after angioplasty.
Bindarit (2-methyl-2-[[1-(phenylmethyl)-1H-indazol-3-yl] methoxy] propanoic acid) is an indazolic derivative, with a specific inhibitory effect on the p65 and p65/p50-induced MCP-1/CCL2 promoter activation15-18. It has been shown to exhibit potent anti-inflammatory activity in a variety of experimental models15-18. In addition, the in vitro models have demonstrated inhibition of smooth muscle cell proliferation, migration and MCP-1 reduction in human coronary arteries by bindarit19. The safety profile and tolerability of the oral formulation of bindarit has previously been demonstrated both in preclinical studies on animal models and in clinical studies on humans15-18. We have also recently demonstrated significant inhibition of in-stent stenosis in porcine coronary arteries following oral administration of bindarit17.
In this explorative trial, we assessed the efficacy and safety of different bindarit doses compared to placebo in preventing restenosis in patients undergoing PCI with BMS (MULTI-LINK VISION®; Abbott Vascular, Santa Clara, CA, USA).
Methods
STUDY DESIGN
This was a pilot phase II, double-blind, placebo-controlled, dose-finding, multicentre, randomised trial in patients requiring clinically indicated coronary intervention. The study was carried out between January 2009 and April 2011 at 20 active sites in Italy, Russia and the Ukraine (ClinicalTrials.gov Identifier: NCT01269242). The sponsor of the clinical trial was ACRAF S.p.A. (Aziende Chimiche Riunite Angelini Francesco), S. Palomba, Pomezia (Rome), Italy, who were responsible “for the initiation, management and financing of the clinical trial” according to the DIRECTIVE 2001/20/EC OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 4 April 2001 (article 2).
ETHICAL APPROVAL
Prior to the study, the protocol, including the informed consent form, was submitted to the local ethics committees. The trial started after obtaining approval from the relevant authorities and local ethics committees. The study was performed in accordance with GCP (CPMP/ICH/135/1995) and the Declaration of Helsinki. Before recruitment, patients were fully informed by the investigators of all aspects of the trial (purposes of the research, possible benefits, any potential personal reasonable risk or discomfort, the expected duration of their participation, as well as procedures and laboratory tests to be undergone). All of this information was comprehensively reported in the information sheets that were given to the patients (enclosed with the informed consent form). If patients agreed to participate, they entered the trial by signing the informed consent form which was countersigned by the investigator.
PATIENT SELECTION
Patients scheduled for elective PCI for documented symptomatic coronary artery disease with a maximum of two de novo lesions (>70% stenosis) were approached to participate in the study. Each lesion should have required a single stent not longer than 28 mm and with a diameter of at least 2.5 mm.
At the baseline visit, all patients underwent procedures to assess their eligibility for the study, which included medical assessment (medical history, cardiovascular risk factors, angina status, bleeding episodes), physical examination, 12-lead ECG, recording of vital signs, and blood and urine sample tests. The urine pregnancy test was performed when appropriate. Strict inclusion and exclusion criteria were applied prior to recruiting (Table 1).
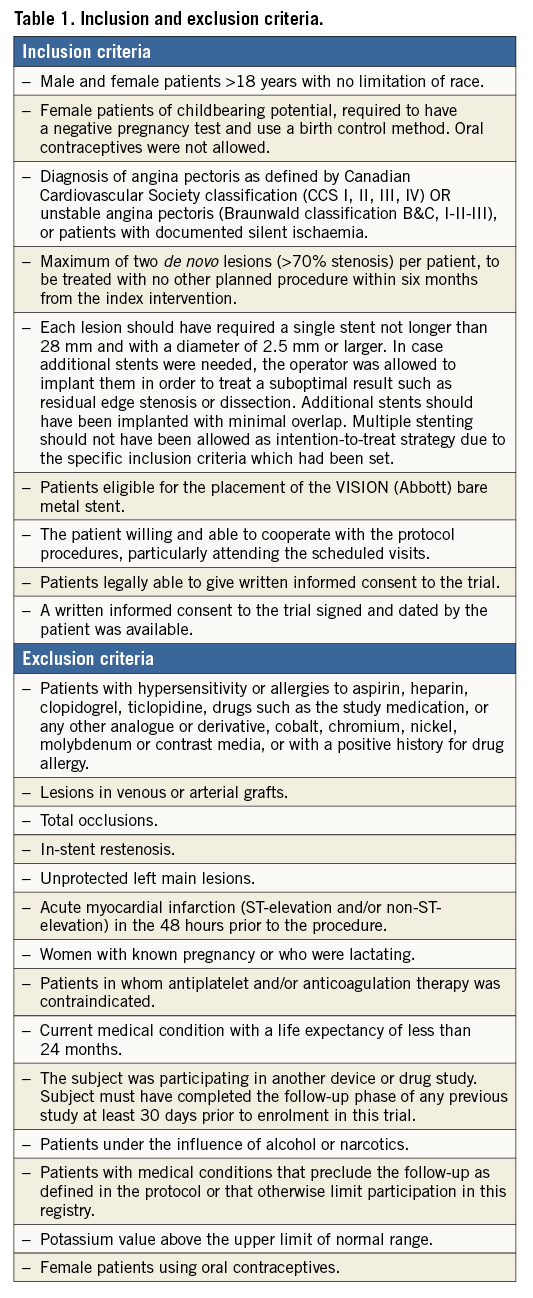
Patients who met the eligibility criteria were assigned to treatment (bindarit 600 mg, bindarit 1,200 mg or placebo) as per the randomisation list provided by the biometric service of the sponsor, and generated by a computer with Microsoft Access 2000. Since this was a double-blind study, neither the investigators nor the patients were aware of the assigned treatment. Bindarit formulation and placebo formulation tablets were indistinguishable in terms of colour and size. The investigators properly directed the assignment of each product, following the sequential order of the specific randomisation list. The study protocol demanded that the investigators balance, as far as possible, the number of diabetic and non-diabetic subjects within each treatment group. This could not be achieved. However, since the randomisation list ensured that equal numbers of diabetic patients were allocated to each treatment group, diabetic status was not a confounder in this study.
PROCEDURE
Patients underwent PCI in accordance with the European Society of Cardiology guidelines on myocardial revascularisation20. All patients in the trial were treated with BMS (MULTI-LINK VISION). The standard dual antiplatelet (aspirin and clopidogrel) therapy was given for at least one month, followed by long-term aspirin treatment. The use of additional drugs such as bivalirudin and GP IIb/IIIa inhibitors was left to the discretion of the operators. The other medications and their posology were recorded for each subject on the case report form and collected in the study database. Additional stents were allowed to treat suboptimal results, such as residual edge stenosis or dissection, but with minimal overlap. Multiple stenting was not allowed as an intention-to-treat strategy due to the pre-specified inclusion criteria.
Following the procedure, before randomisation, all patients had their blood samples taken for MCP-1/CCL2 levels.
TRIAL DRUG ADMINISTRATION
Two different doses of bindarit (600 mg and 1,200 mg daily) were investigated in the study. Phase I clinical studies showed that bindarit is well tolerated in healthy volunteers (up to 1,200 mg twice daily orally, for 12 days), confirming the good tolerability profile. In addition, several phase II clinical trials have been performed in patients with rheumatic disease, rheumatoid arthritis, hypertriglyceridaemia and hypoalphalipoproteinaemia21,22. In these studies, bindarit was orally administered for periods of up to six months and different doses were used, from 300 mg up to 1,200 mg. In our trial, two different bindarit doses were investigated, 600 mg and 1,200 mg daily. Assuming a significant effect of the 1,200 mg daily dose, it was important to test whether a lower dose (600 mg daily) was equally effective. Indeed, the same effect with a reduced patient exposure to the study drug was expected to increase both the risk/benefit and the cost/effectiveness profile of bindarit treatment in preventing restenosis in patients undergoing coronary intervention. Higher doses of bindarit (i.e., 2,400 mg daily) were excluded since, in a previous trial on patients with rheumatoid arthritis, a higher dose resulted in an increased incidence of adverse events per patient (in particular, gastrointestinal reactions). Taking into account that patients recruited to this study were frequently treated with several concomitant therapies, it was considered not ethically correct to overexpose patients to a higher risk of adverse events occurrence. After the procedure, eligible patients were randomised into three arms: bindarit 600 mg, bindarit 1,200 mg or bindarit placebo. Two tablets (each containing 300 mg of bindarit or placebo according to the selected doses) were administered orally twice daily.
All study medications (manufactured by Angelini-ACRAF, Rome, Italy) were identical in appearance to maintain blinding. The first dose of the assigned medication was administered within three hours of the PCI. The patient was considered evaluable if the compliance rate was at least 80%.
FOLLOW-UP
Patients were followed up regularly (one, 30, 60, 90, 120, 150 and 180 days after PCI). During each follow-up visit, patients underwent medical assessment and were evaluated for angina, major adverse cardiac events (MACE) and bleeding episodes. Patients were asked about concomitant treatments and the occurrence of any adverse events. Blood and urine samples were collected for detailed laboratory analyses including plasma MCP-1/CCL2 levels. At 180 days, patients were advised to stop the test medication and all patients underwent control coronary angiography. Coronary angiography was also performed earlier, if clinically indicated. At 270-day clinical follow-up, subjects underwent medical assessment and enquiry about adverse events. Blood samples were collected to measure only MCP-1/CCL2 levels during this final visit. MCP-1/CCL2 levels were measured in a centralised laboratory (Research Laboratory in Immunology and Inflammation of the Istituto Clinico Humanitas-IRCCS, Rozzano, Milan, Italy) by an independent analyst blinded to treatment allocation, using commercially available kits (catalogue numbers: DY282 and SCP00; R&D Systems Europe, Ltd., Abingdon, United Kingdom). For MCP-1/CCL2, the limit of detection was 60 pg/ml.
Standard QCA analysis was performed on the angiograms obtained before (QCA-pre) and immediately after the PCI (QCA-post) and at six-month follow-up on control angiogram (QCA-fu) by an independent core laboratory (Mediolanum Cardio Research S.r.l., Cinisello Balsamo, Milan, Italy).
All MACE were reviewed and adjudicated by an independent clinical events committee according to ARC criteria23.
STUDY ENDPOINTS
The primary endpoint of the study was in-segment late loss (in-stent and 5 mm proximal and distal to the stent) measured by QCA at six months from the index procedure. The secondary endpoints were in-stent late loss measured by QCA at six months, binary angiographic restenosis, MACE at nine months, the assessment of MCP-1/CCL2 levels, and the safety profile of bindarit.
Late luminal loss was defined as the difference between MLD in QCA-post and MLD in the QCA-fu. Binary angiographic restenosis was defined as >50% luminal narrowing at the segment site (stent and 5 mm proximal and distal) on follow-up QCA, regardless of clinical symptoms.
MACE was defined as the combination of cardiac death, MI and TVR. Deaths were classified as either cardiac or non-cardiac. Cardiac death was defined as any death due to a cardiac cause, procedure-related death and death from unknown cause.
Efficacy assessment was planned for two study populations:
– Intention-to-treat (ITT), defined as all randomised patients who received at least one dose of the experimental drugs, who had complete baseline data and QCA follow-up assessment.
– Per-protocol (PP), defined as all randomised patients who met eligibility criteria, had adequate compliance (at least 80%) to the study treatment, received aspirin+clopidogrel as dual antiplatelet therapy and who were required to have a follow-up angiogram under strict temporal criteria. Patients who underwent QCA follow-up before 30 days or after 270 days from the index procedure were not included in the PP population. Patients who underwent QCA follow-up between the 31st and 89th day after the index procedure were included in the PP population only when the study endpoint was reached (restenosis in the target lesion).
Safety and tolerability assessment were performed on all patients who received at least one dose of the allocated intervention irrespective of QCA follow-up data.
SAMPLE SIZE CALCULATION AND STATISTICAL ANALYSIS
Considering that this was a preliminary exploratory study, no specific sample size calculation was performed. In order to obtain an acceptable representation, we planned to enrol 120 evaluable subjects divided into three groups.
Continuous data were expressed as mean±standard deviation, and categorical data as proportions (%). Comparisons of clinical and angiographic characteristics of patients were performed using the Student’s t-test. The categorical variables were compared using the chi-square test. A one-way ANOVA for independent groups was conducted to compare the effect of bindarit on in-stent late loss and in-segment late loss in BND 600, BND 1,200 and placebo treatment groups. All analyses were conducted using SPSS software, Version 18.0 (SPSS Inc. Chicago, IL, USA), and all reported p-values are two-sided. Values of p<0.05 were regarded as statistically significant.
Results
The results from the study were presented at ACC 2012. One hundred and fifty-two patients were screened, but three patients were excluded prior to randomisation as they did not meet the inclusion criteria or withdrew consent from the study. The remaining 149 patients were randomised into three treatment arms: 48 in the BND 600 mg group, 50 in the BND 1,200 mg group and 51 in the placebo (PLB) group. One patient in the BND 1,200 mg group did not take the allocated treatment and was excluded. Demographic and clinical characteristics of the randomised patients at screening are summarised in Table 2. There were no significant differences in the baseline characteristics among the three treatment groups. Thirty-seven out of 148 patients included in the safety population had diabetes: 12 in the BND 600 mg group, 11 in the BND 1,200 mg group and 14 in the PLB group.
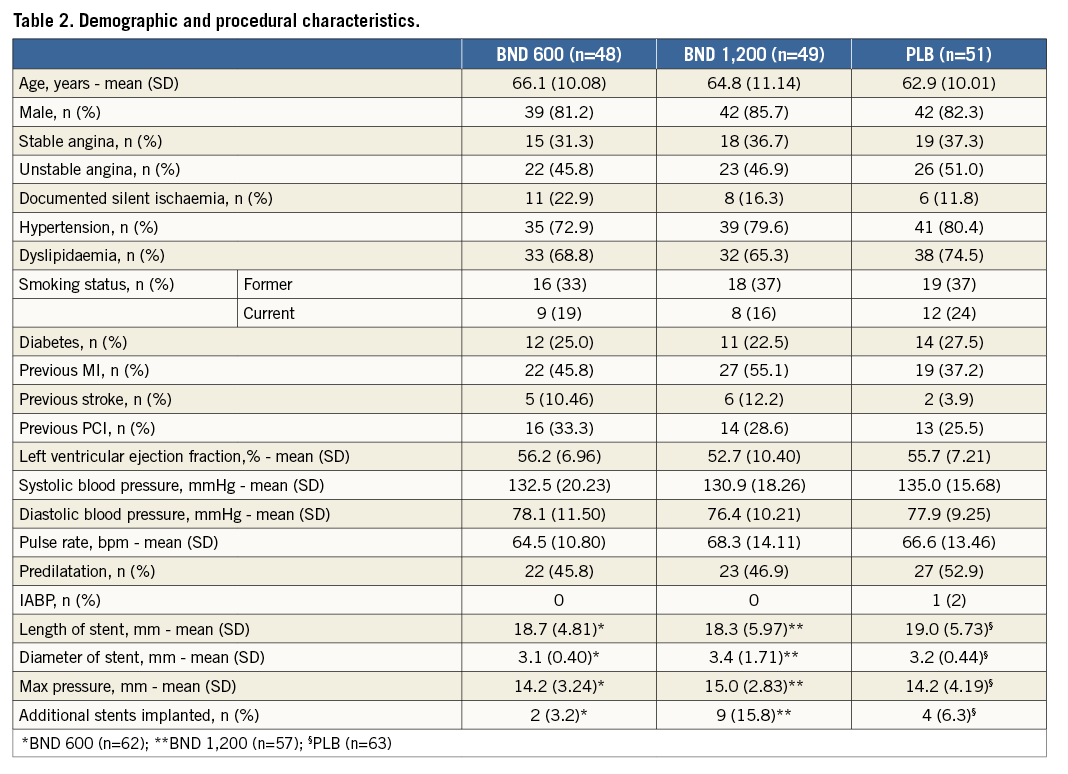
Angiographic and index procedural characteristics by treatment arm are described in Table 2. No significant differences were observed in procedural characteristics and complications among the treatment groups. The overall compliance to bindarit and placebo tablets was >90% (96% in the BND 600 group, 97.2% in the BND 1,200 group and 92% in the PLB group).
ANGIOGRAPHIC FOLLOW-UP AT SIX MONTHS
Efficacy analysis was carried out on two study populations (Figure 1):
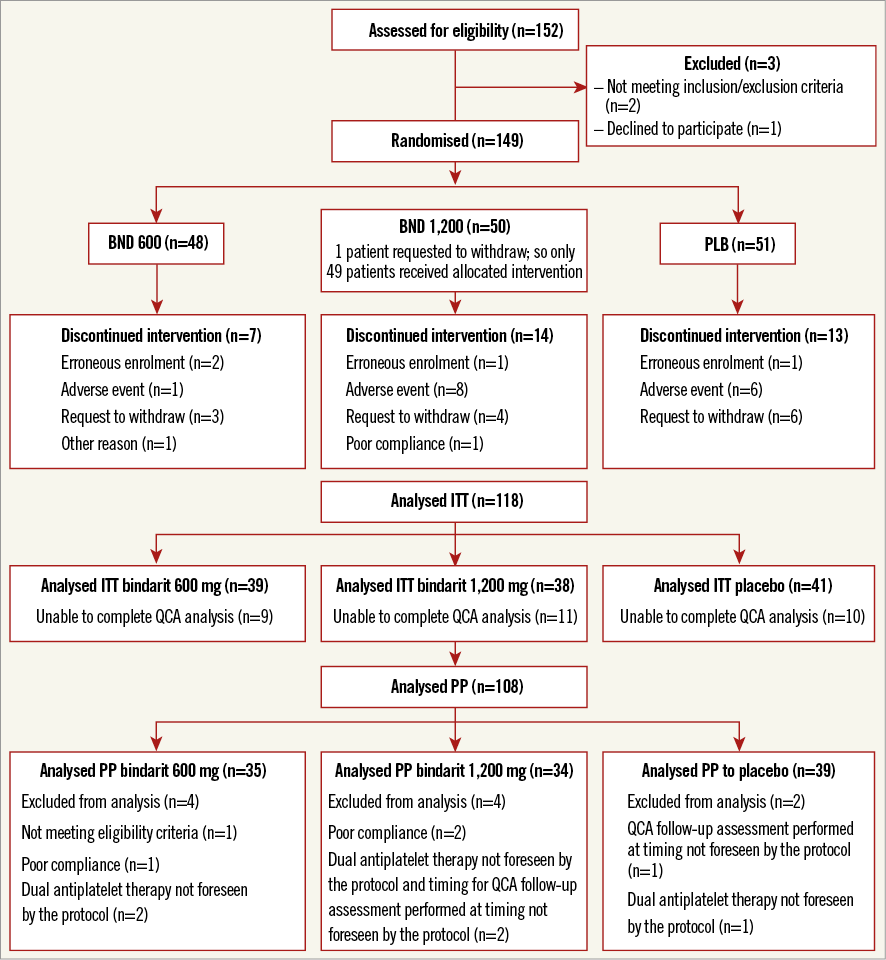
Figure 1. CONSORT flow diagram showing the number of patients recruited in each of the study populations.
– ITT population comprising 118 patients. Thirty patients out of a total of 148 randomised patients were excluded from the ITT population, as the follow-up QCA analysis was not available.
– PP population comprising 108 patients. The PP population was derived by excluding 10 patients from the ITT population due to use of different antiplatelet agents or timing of QCA not within the initial set protocol or unable to complete the intended treatment with adequate compliance (at least 80%).
PRIMARY ENDPOINT (IN-SEGMENT LATE LOSS)
The details of QCA analysis before and after stenting and at six-month follow-up in the ITT and PP populations are provided in Table 3. In-segment late loss was numerically higher in the placebo arm compared to the bindarit groups (both BND 600 and BND 1,200 mg) at follow-up and was more evident in the PP than in the the ITT population. However, both failed to reach statistical significance (p=0.12 and p=0.21, respectively) (Figure 2A, Figure 2B).
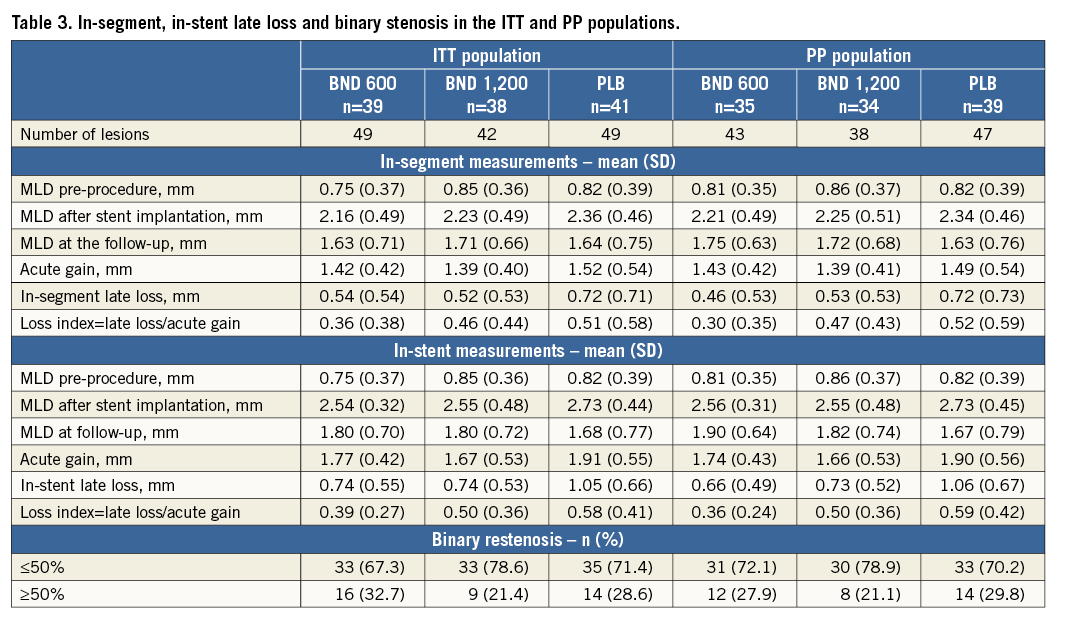
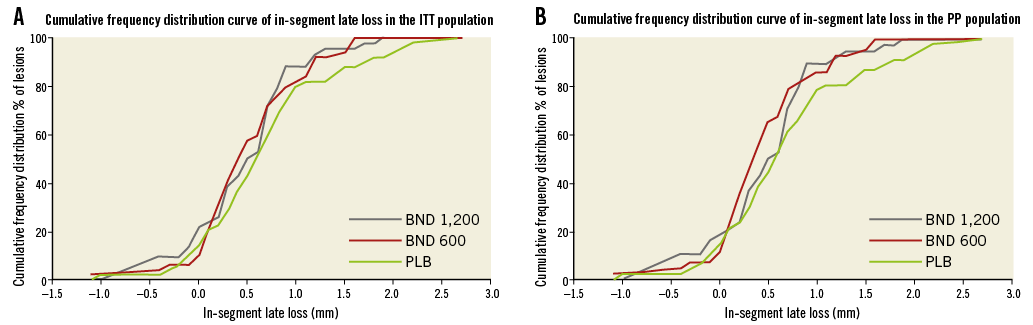
Figure 2. Cumulative frequency distribution curves of in-segment late loss among the three treatment arms in the ITT population (A) and in the PP population (B).
SECONDARY ENDPOINT
IN-STENT LATE LOSS
The details of MLD measurements before and after stenting and at six-month follow-up, acute gain, late loss and late loss index in the ITT and PP populations are provided in Table 3. In-stent late loss was significantly higher in the placebo arm compared to the bindarit groups in both the ITT and PP populations (p=0.01 and p=0.003, respectively) (Figure 3A, Figure 3B). This difference was significantly greater in the PP than in the ITT population.
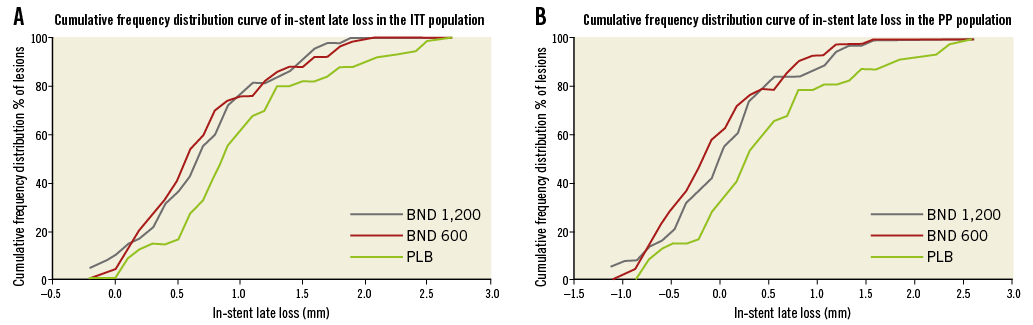
Figure 3. Cumulative frequency distribution curves of in-stent late loss among the three treatment arms in the ITT population (A) and in the PP population (B).
BINARY RESTENOSIS
The incidence of binary restenosis was not significantly different between the treatment arms in the ITT and the PP populations, though numerically it was higher in the BND 600 group as compared to the PLB group in the ITT population (Table 3). The patterns of restenosis were: diffuse (BND 600: six [40%], BND 1,200: five [55.6%], PLB: eight [57.1%]) and focal (BND 600: six [53.3%], BND 1,200: four [44.4%], PLB: four [28.6%]). In the ITT population, one lesion in the BND 600 group and two lesions in the PLB group were occlusive restenosis. In the PP population, two lesions in the placebo group were occlusive restenosis and there were none in the bindarit group.
MACE
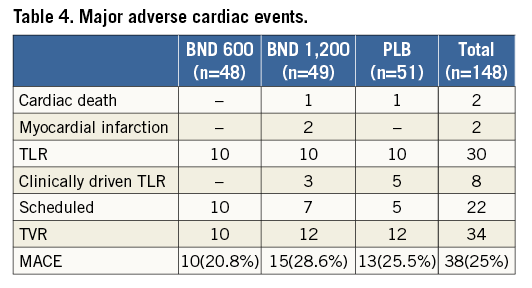
The clinical events committee evaluated forty-nine events (14 BND 600, 19 BND 1,200 and 16 PLB). Thirty-eight (10 BND 600, 15 BND 1,200 and 13 PLB) out of 49 events were judged as MACE. No significant differences in the incidence of MACE at six months were found among treatment groups (20.8% in BND 600 mg vs. 28.6% in BND 1,200 mg vs. 25.5% in PLB, p=0.54) (Table 4). However, the incidence of MACE in the BND 600 mg group tended to be numerically lower. There were no cardiac deaths, MI or clinically driven TLR and TVR in the BND 600 mg group.
INFLAMMATORY BIOMARKERS
The plasma levels of MCP-1/CCL2 were measured at baseline and at the follow-up visits and are graphically depicted in Figure 4. There were no differences in the plasma MCP-1/CCL2 levels between the treated and untreated patients at any time point. In addition, there was no correlation between MCP-1/CCL2 levels and either in-segment late loss or in-stent late loss.
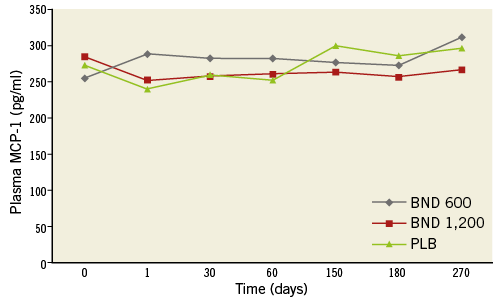
Figure 4. Graphical depiction of plasma levels of MCP-1/CCL2 measured at post PCI and at follow-up among the three treatment arms.
ADVERSE EVENTS
During the study, 371 adverse events (AE) were reported by 112 patients: 357 out of 371 were reported after the initiation of the treatment. The summary of AE is shown in Table 5. The incidence of AE, especially serious and severe in intensity, was similar in the BND 600 mg and PLB groups. Patients treated with BND 1,200 mg had an overall greater number of AE compared to the other two groups (BND 1,200=36%; BND 600=30%; PLB=34%). Eighty-three serious AE occurred during the trial, but only five were judged to be treatment-related. There were three cases of mild-to-moderate hyperkalaemia (one in each group), one case of cholestatic hepatitis and one case of transaminase elevation (both in the BND 1,200 mg group). Two deaths occurred in the trial, one in the placebo arm and one in the BND 1,200 mg arm. Both patients were not under treatment at the time of death. Within a generally satisfactory tolerability profile, bindarit 1,200 mg appeared to be the least tolerated.

Discussion
The main findings of this study are:
– Bindarit showed a trend towards a reduction in in-segment late loss, but this did not reach statistical significance.
– Bindarit significantly reduced in-stent late loss after stenting compared to placebo.
– Bindarit was well tolerated, with over 90% compliance and without any major side effects compared to placebo.
The bindarit study failed to meet the primary endpoint, although the trends in the reduction of in-segment late loss were higher in the bindarit groups than in the placebo. The significant reduction in in-stent late loss in the bindarit groups is interesting, indicating that bindarit may exert a favourable action on the vessel wall following stent implantation. Targeting a specific patient cohort vulnerable to ISR might produce clinically significant outcomes. In addition, the neutrophil and monocyte recruitment to the vessel wall is observed within a few hours of balloon injury to the vessel wall6,7,24,25. In this trial, the patients were loaded with bindarit after PCI. It might therefore have taken longer to achieve the steady state of the drug and hence it may have been too late to influence the inflammatory cascade initiated by endothelial denudation and platelet deposition that promotes leukocyte recruitment. Loading the drug a few days before PCI might increase its antiproliferative effects. As expected, the effects of bindarit were more pronounced in the PP population who completed the duration of treatment than in the ITT population. Bare metal stents were used in this trial as we feel that the use of bindarit will predominantly be in patients who are not eligible for drug-eluting stents, and the incidence of restenosis is relatively higher in BMS.
The lack of a linear relationship between late loss and MCP-1/CCL2 plasma levels and, more importantly, the absence of differences in MCP-1/CCL2 levels among the treatment groups raise questions about the exact mechanism of action of bindarit in this setting. Our preclinical study on porcine coronary arteries had demonstrated significantly high levels of MCP-1/CCL2 in pigs which underwent PCI. The pigs which were treated with bindarit had significant reduction in the MCP-1/CCL2 plasma concentration and intimal hyperplasia inhibition17. Most studies demonstrating reduction of MCP-1/CCL2 levels using bindarit have been performed in animal models15-18. There are only limited studies which have studied bindarit in humans and none of them has measured plasma MCP-1/CCL2 levels. The only clinical data where the reduction in MCP-1 was evidenced were obtained in lupus nephritis, where the treated patients showed a significant reduction of MCP-1 excreted in urine26. The other possible explanation may be related to the plasma levels of MCP-1/CCL2, which may dictate the suppression by bindarit. It may be that not all patients undergoing PCI have elevated MCP-1/CCL2 levels, and bindarit may not have a significant effect in patients in whom the MCP-I/CCL2 levels are not elevated following PCI. This may also be the reason why some patients are more vulnerable to restenosis. From our current knowledge, we do not know or cannot predict which patients are more prone to have elevation in their MCP-1/CCL2 levels. Further studies are warranted specifically to study the mechanism of action of bindarit before drawing any meaningful conclusions.
Previous studies which have evaluated oral antirestenotic agents (oral rapamycin and oral steroids) have found some benefits with these agents, but none of them has translated into routine clinical practice. In the ORAR II study, 100 patients were randomised to either oral rapamycin or placebo27. At nine months, patients who received rapamycin had significantly lower in-segment late loss (0.66 mm) as compared to placebo (1.13 mm). This value (0.66 mm) is higher when compared to our study where the in-segment late loss was 0.46 mm in the BND 600 mg group. However, the late loss was also lower in patients who received BMS, which may be due to several factors (procedural characteristics, stent design, patient and lesion characteristics). In the CEREA-DES study, the effect of oral steroids was assessed28. Patients (n=375) were randomised to BMS, BMS+oral steroids or DES. Only clinical endpoints were measured in this trial. The trial demonstrated that patients receiving BMS had significantly lower event-free survival (CV death, MI and TVR) as compared to the other two groups. Although all these trials including ours are encouraging, we need more robust data with a larger group of patients before these treatments translate into routine clinical practice. The availability of second- and third-generation drug-eluting stents may make these findings clinically not relevant. Nevertheless, there are several patients who may not be able to tolerate or safely assume dual antiplatelet therapy for an extended period of time. In these patients, the search for an acceptable substitute to limit intimal hyperplasia after stenting may appear a reasonable endeavour. In addition, indications for PCI are escalating and will continue to do so. Embarking on complex lesions (diffuse and calcified lesions, bifurcations, chronic total occlusions, restenotic lesions) in high-risk patients (diabetes, chronic kidney disease) will invariably produce restenosis. None of the current DES is immune to it. In these patients, the search for a complementary technology to limit intimal hyperplasia may appear a reasonable endeavour.
Bindarit was reasonably well tolerated with compliance of >90% with no significant life-threatening adverse effects. Although most patients tolerated bindarit reasonably well, the incidence of side effects and adverse events was slightly higher in the BND 1,200 group. In addition, none of the patients in the BND 600 mg arm underwent clinically driven TLR or TVR. These findings suggest that bindarit at a dose of 600 mg is probably sufficient to produce the intended benefit and is relatively better tolerated.
Limitations
The larger follow-up MLD explained by a smaller late loss in the bindarit groups did not translate into a better clinical outcome for these patients, which might be explained by the small sample size and by the small reduction in late loss. Nevertheless, the study was not powered to evaluate the clinical endpoints.
Conclusions
Bindarit failed to reduce in-segment late loss significantly, although the trends were in favour of bindarit rather than placebo. However, in-stent late loss was significantly less in the bindarit group as compared to the placebo group. Bindarit was well tolerated, with a compliance rate of over 90% with no major adverse events. An increased dose of bindarit did not offer any advantage, but in fact the high dose was relatively less well tolerated.
| Impact on daily practice Bindarit has demonstrated trends in the reduction of intimal hyperplasia following coronary angioplasty. Bindarit may be considered in patients who are at high risk of restenosis (diabetes, chronic kidney disease, previous restenosis, etc.), although further evidence with studies evaluating an earlier timing of administration is needed to reinforce these findings. |
Conflict of interest statement
The authors have no conflicts of interest to declare.
