
Abstract
Aims: Treatment of in-stent restenosis of coronary stents is challenging. The use of drug-coated balloons (DCB) is a promising technique to treat in-stent restenosis without adding another metal layer. The aim of the AGENT ISR randomised trial is to evaluate angiographic and clinical outcomes in patients with ISR of a previously treated lesion who were treated with either a DCB with a new coating formulation (Agent) or a standard DCB (SeQuent Please).
Methods and results: AGENT ISR is a multicentre, randomised, open-label, non-inferiority study comparing the Agent and SeQuent Please DCB. A total of 125 patients (mean age ~68 years, 18% female) with in-stent restenosis of a previously treated lesion <28 mm in length were randomised at 11 sites in Europe to Agent (n=65) or SeQuent Please (n=60). The primary endpoint, six-month in-stent late lumen loss, in the Agent group (0.397±0.43 mm [n=51]) was non-inferior to that of the SeQuent Please group (0.393±0.536 mm [n=49]), as the two-sided upper 95% confidence boundary for the difference between groups was less than the pre-specified non-inferiority margin of 0.20 (difference 0.004, 95% CI [−0.189, 0.196]; pnon-inferiority=0.046). At one year, mortality was 3.1% in Agent and 1.7% in SeQuent Please patients (p>0.99), target lesion revascularisation 7.7% versus 10.0% (p=0.89), and stent thrombosis 0% versus 3.3% (p=0.44). Similar improvements in quality of life were seen in the two groups.
Conclusions: In this head-to-head comparison of two DCB, Agent proved to be non-inferior to SeQuent Please for in-stent late lumen loss at six months. Clinical Trials Registration: NCT02151812 (http://clinicaltrials.gov/)
Introduction
Drug-eluting stents (DES) are the treatment of choice for most patients with coronary artery disease. Restenosis is significantly reduced with DES compared to bare metal stents (BMS) or balloon angioplasty1. Even with DES, in-stent restenosis (ISR) requiring revascularisation may occur in up to 10% of patients1. There are several treatment options for ISR, including balloon angioplasty, cutting balloons, DES, and drug-coated balloons (DCB). DCB components include a balloon, an antiproliferative drug, and the carrier substance or excipient2. The balloon is inflated at the target site and the drug-excipient mixture is deposited onto the arterial surface, allowing transfer into the vessel wall. Paclitaxel is used on most DCB due to its lipophilicity, absorption, and retention in the vessel wall. In preclinical testing, the transfer of antirestenotic drug to the tissue was influenced by the presence and type of excipient3,4. There are several theoretical benefits of DCB over stents. The most important represents treatment without a permanent vascular implant; additional metal layers in a coronary artery can impart a higher risk of stent thrombosis5. Several studies have shown beneficial results with DCB in patients with BMS-ISR or DES-ISR, usually in comparison to balloon angioplasty alone or DES6,7,8,9,10,11,12.
The aim of the AGENT ISR randomised trial is to evaluate angiographic and clinical outcomes in patients with ISR of a previously treated lesion who were treated with either a DCB with a new coating formulation (Agent) or a standard DCB (SeQuent Please).
Methods
STUDY DESIGN AND PATIENT SELECTION
AGENT ISR is a multicentre, randomised, open-label, non-inferiority study comparing two paclitaxel-coated balloons for coronary in-stent restenosis treatment at 11 European sites (Supplementary Appendix 1). The Agent DCB (co-developed by Hemoteq AG, Würselen, Germany, and Boston Scientific, Marlborough, MA, USA) is coated with a formulation of paclitaxel and a highly efficient excipient acetyl tri-butyl citrate (paclitaxel 2 μg/mm2) and was compared to the SeQuent® Please DCB (B. Braun Melsungen AG, Berlin, Germany; 3 μg/mm2 paclitaxel in an iopromide matrix).
Independent ethics committees at each centre approved the study protocol. All patients provided written informed consent before enrolment. All clinical events were adjudicated by an independent clinical events committee. An independent core laboratory evaluated angiograms (CoreLab Black Forest GmbH, Bad Krozingen, Germany). The study is registered at www.clinicaltrials.gov (identifier: NCT02151812).
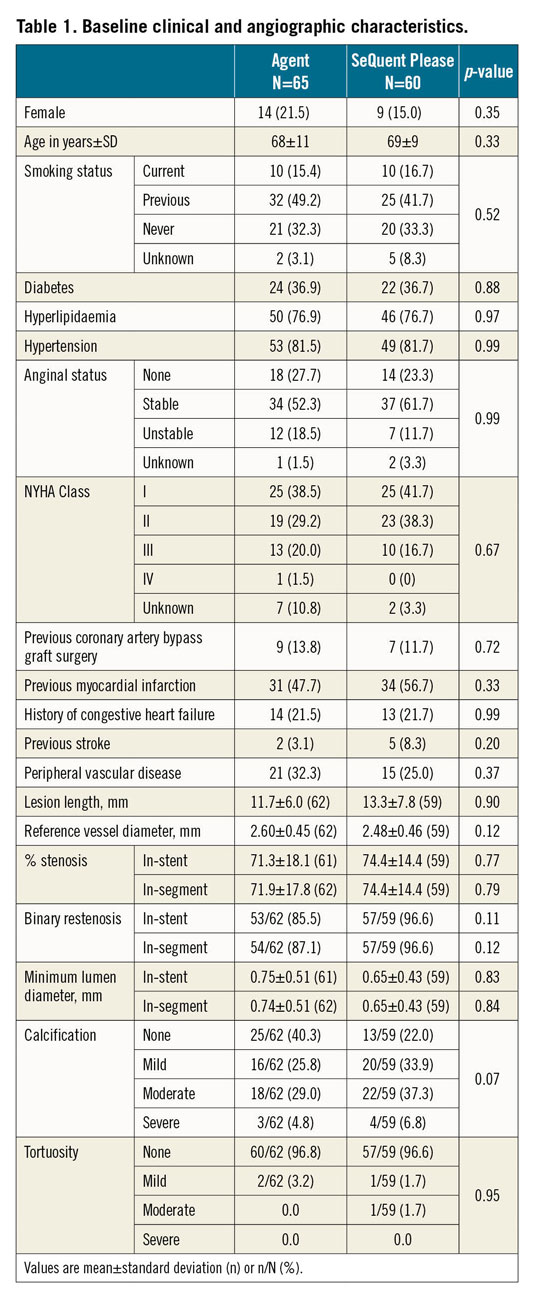
Eligible patients were ≥18 years old with ISR of a previously treated (DES or BMS) native coronary artery lesion of up to 28 mm in length (by visual estimate) in a native coronary artery with a diameter of 2.0 mm to 3.5 mm. The target lesion had to be coverable with one DCB. Patients with left main (LM) disease, recent or planned percutaneous coronary intervention (PCI) or coronary artery bypass graft surgery (CABG), chronic total occlusion (CTO), or acute/recent myocardial infarction (MI) were excluded. Subjects who satisfied study selection criteria were randomly assigned 1:1 to receive treatment with Agent or SeQuent Please DCB after informed consent had been obtained and the target lesion was successfully predilated with an uncoated angioplasty balloon. Randomly permuted blocks with random block sizes (sampled from a discrete uniform distribution) were employed to ensure approximate balance of treatment allocation, and all study centres received an independent randomisation sequence which was stored in opaque envelopes with an independent randomisation sequence. Every centre received a stack of envelopes with its randomisation sequence before the patient recruitment started. Dual antiplatelet therapy was recommended for three months after the index procedure, followed by acetylsalicylic acid monotherapy. Clinical follow-up will continue to three years post procedure. An angiographic follow-up was conducted at six months after the index procedure. Post-procedural and six-month follow-up visits occurred in person, whereas 30-day and annual visits were conducted either in person or via the telephone.
INTERVENTIONAL PROCEDURE
Test devices (Agent) were 8-30 mm in length with a diameter of 2.00-3.50 mm; control devices (SeQuent Please) were 10-30 mm with a diameter of 2.00-3.50 mm.
Patients were randomised 1:1 after informed consent had been obtained from the patient and after successful predilatation of the target lesion. The investigator treated the target lesion with the assigned DCB. One DCB was allowed per lesion with one planned inflation as transfer of consistent therapeutic drug levels occurs only during the first inflation. Repeat inflation of DCB at the treatment site was limited to emergency/bail-out situations. Investigators were instructed to follow the instructions for use for each device. The use of adjuvant therapies (i.e., rotablation, laser atherectomy, cutting balloon, and other drug-coated balloon) within the target vessel was not allowed according to the protocol. Investigators were instructed to follow the instructions for use for each device.
ENDPOINTS
The primary endpoint of the present study was in-stent late lumen loss, which was defined as the difference between minimal lumen diameter of the target vessel after the index procedure and at six months, evaluated by quantitative coronary angiography (QCA). QCA was assessed by an independent, blinded angiographic core laboratory (CoreLab Black Forest GmbH). Technical success was defined as the ability to cross and dilate the lesion to achieve residual angiographic stenosis <30%. Clinical procedural success was defined as technical success with no incidence of death/MI within 24 hours of the procedure. Clinical endpoints were evaluated in-hospital, at 30 days, six months, and annually to three years and included death, MI (third universal definition)13, target lesion revascularisation (TLR) and target vessel revascularisation (TVR). Stent thrombosis was defined according to Academic Research Consortium (ARC) criteria14, as the occurrence of definite stent thrombosis (angiographic confirmation of stent thrombosis, with the presence of a thrombus that originates within the stent or in the segment 5 mm proximal or distal to the stent), or probable stent thrombosis (any unexplained death within the first 30 days after PCI, irrespective of the time after the index procedure, any MI that is related to documented acute ischaemia in the territory of the implanted stent without angiographic confirmation of stent thrombosis and in the absence of any other obvious cause). All major adverse events were adjudicated by a clinical events committee. Additional six-month angiographic parameters included diameter stenosis, binary restenosis, and minimum lumen diameters. Health status was evaluated with the SF-1215 and EQ-5D16 questionnaires at baseline and six and 12 months post procedure.
STATISTICAL ANALYSIS
The study primary endpoint, powered for non-inferiority, was six-month in-stent late lumen loss. Expected late lumen loss was between 0.2 mm and 0.4 mm for both groups with a common standard deviation of 0.3 mm (from the PACCOCATH ISR I, I&II, and PEPCAD II and PEPCAD DES trials6,7,8,17). With a non-inferiority margin of 0.2 mm and 90% power, 122 randomised subjects were required (estimate 10% attrition due to patients lost to follow-up and 10% due to unevaluable QCA data). If the two-sided upper 95% confidence boundary for the difference in six-month in-stent late lumen loss (Agent – SeQuent Please) was less than the pre-specified margin, non-inferiority would be met. This corresponds to p<0.05 from a two-sided t-test comparing the difference between groups to the non-inferiority margin. Continuous variables were estimated as mean±standard deviation and compared with the Student’s t-test. Discrete variables were reported as counts and percentages; differences were assessed by means of the chi-square or Fisher’s exact test.
Results
Between August 2014 and May 2016, 125 patients were randomised 1:1 at 11 sites in Germany and France. A total of 65 patients were allocated to the Agent group and 60 patients to the SeQuent Please group (Figure 1). There was no crossover between the treatment groups. Six-month angiographic follow-up data were available in 78% of Agent and 82% of SeQuent Please patients; one-year clinical follow-up data were available in all patients. Baseline clinical and angiographic characteristics were similar between groups (Table 1, Table 2). The mean patient age was 68 years in Agent patients and 69 years in SeQuent Please patients; women accounted for 21% of Agent and 15% of SeQuent Please patients; 37% of patients in each group had diabetes (Table 1). Technical success was high in both arms (Agent 98.5% vs SeQuent Please 96.7%, p=0.94) as was clinical procedural success (98.5% vs 95.0%, respectively; p=0.56) (Table 2). Post-procedural in-stent binary restenosis was found in 6.5% of Agent and 6.8% of SeQuent Please patients (p>0.99) (Table 2). There are no specific data available on the types of balloon used for predilatation in the present study.
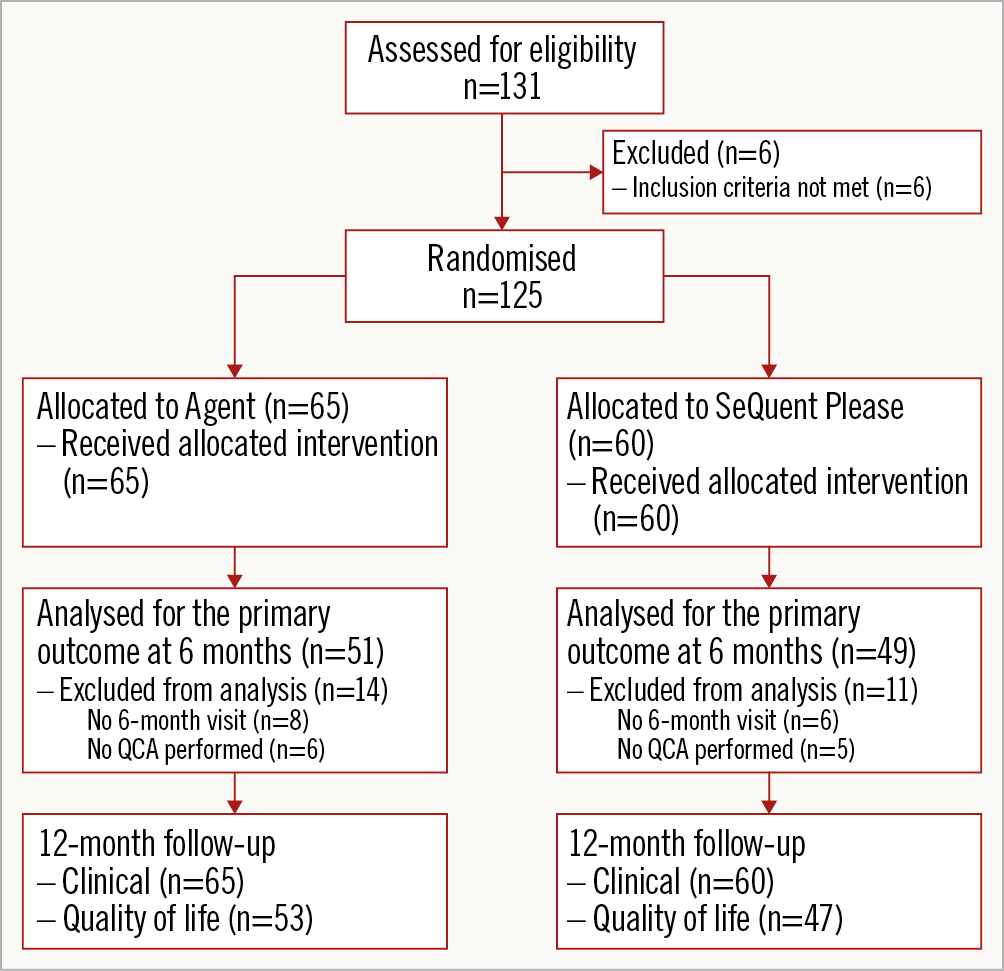
Figure 1. Patient flow and disposition.
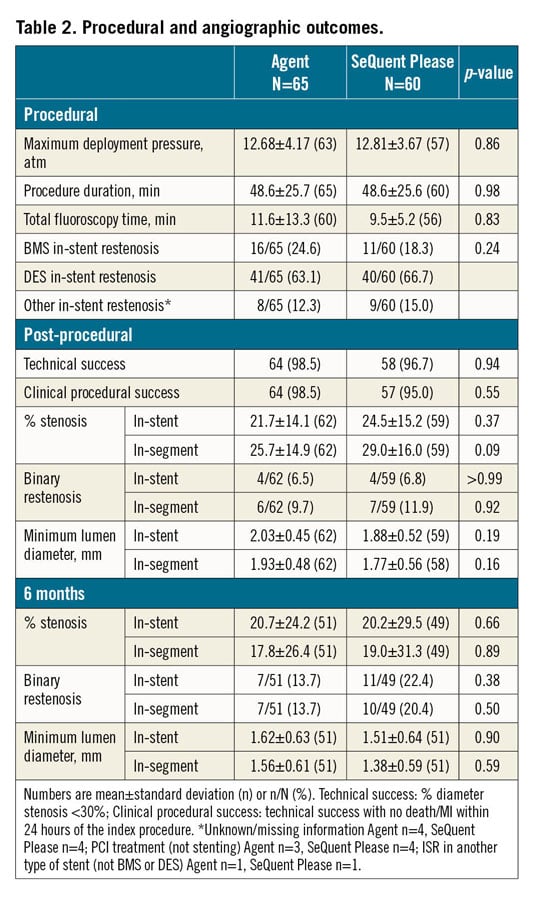
The primary endpoint, six-month in-stent late lumen loss, was 0.397±0.43 mm in the Agent group and 0.393±0.536 mm in the SeQuent Please group (Figure 2). The difference between groups was 0.004 and the upper 95% confidence boundary for the difference between groups was 0.196 (95% CI: −0.189, 0.196), demonstrating non-inferiority (p for non-inferiority=0.046) (Figure 2). Cumulative distribution of acute lumen gain and late lumen loss was similar between groups (Figure 2). Other angiographic parameters at six months were not significantly different between groups (Table 2).
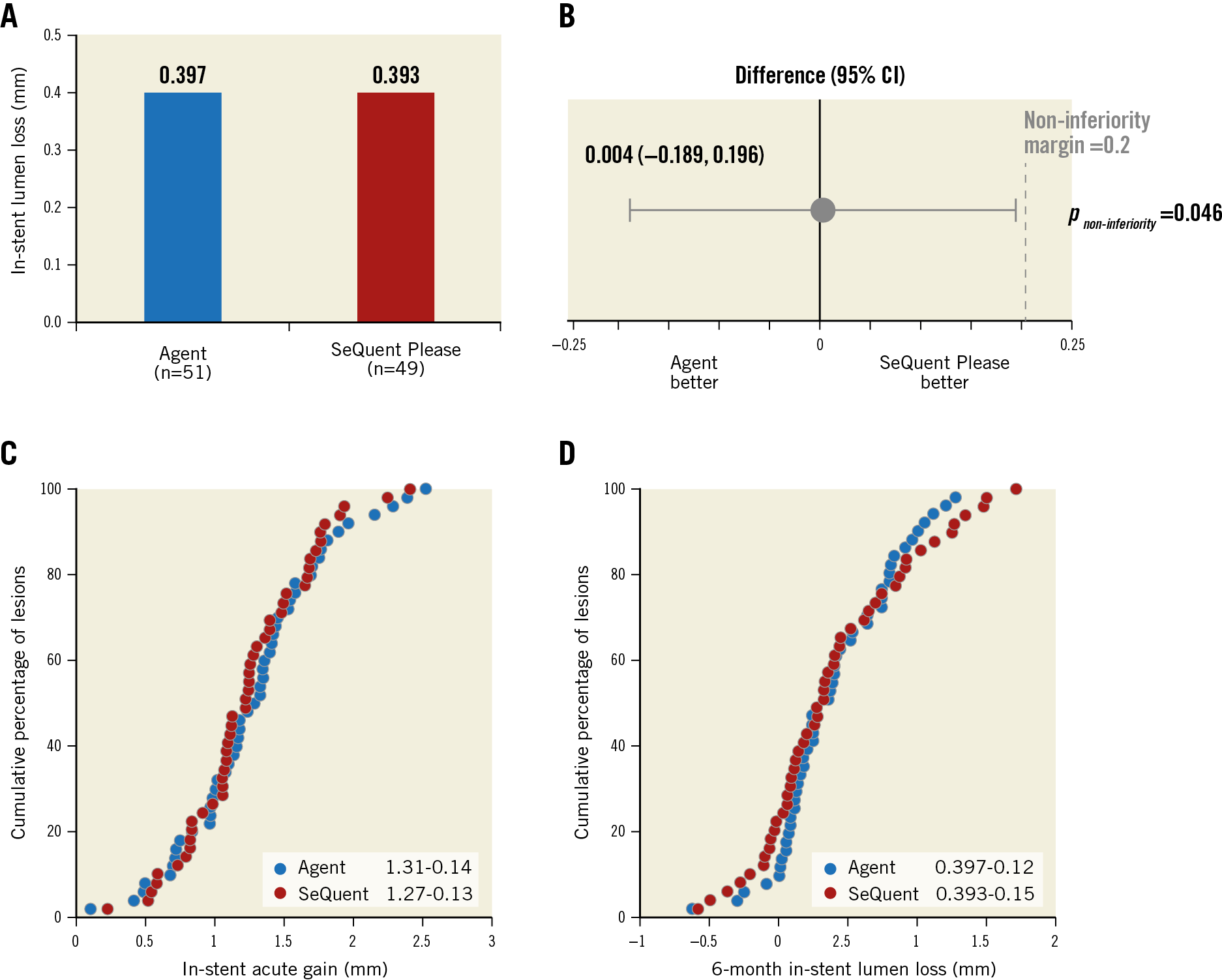
Figure 2. Primary endpoint: six-month in-stent late lumen loss. A) Late lumen loss in Agent (blue) and SeQuent Please (red) cohorts. B) Difference in late lumen loss; error bars indicate two-sided 95% confidence intervals. C) & D) Cumulative frequency distribution curves of lesion-level in-stent acute gain (C) and in-stent late lumen loss (D) at six months.
At one year, death occurred in 3.1% (2/65) of Agent versus 1.7% (1/60) of SeQuent Please patients (p>0.99) (Figure 3). All deaths were considered cardiac-related. MI occurred in 4.6% of Agent versus 3.3% of SeQuent Please (p>0.99) patients, TLR in 7.7% versus 10.0% (p=0.89), TLF in 12.3% versus 11.7% (p>0.99), and stent thrombosis in 0.0% versus 3.3% of patients (p=0.44) (Figure 3). The two DCB were associated with similar improvements in quality of life, as assessed using the SF-12 and EQ-5D questionnaires (Supplementary Table 1).
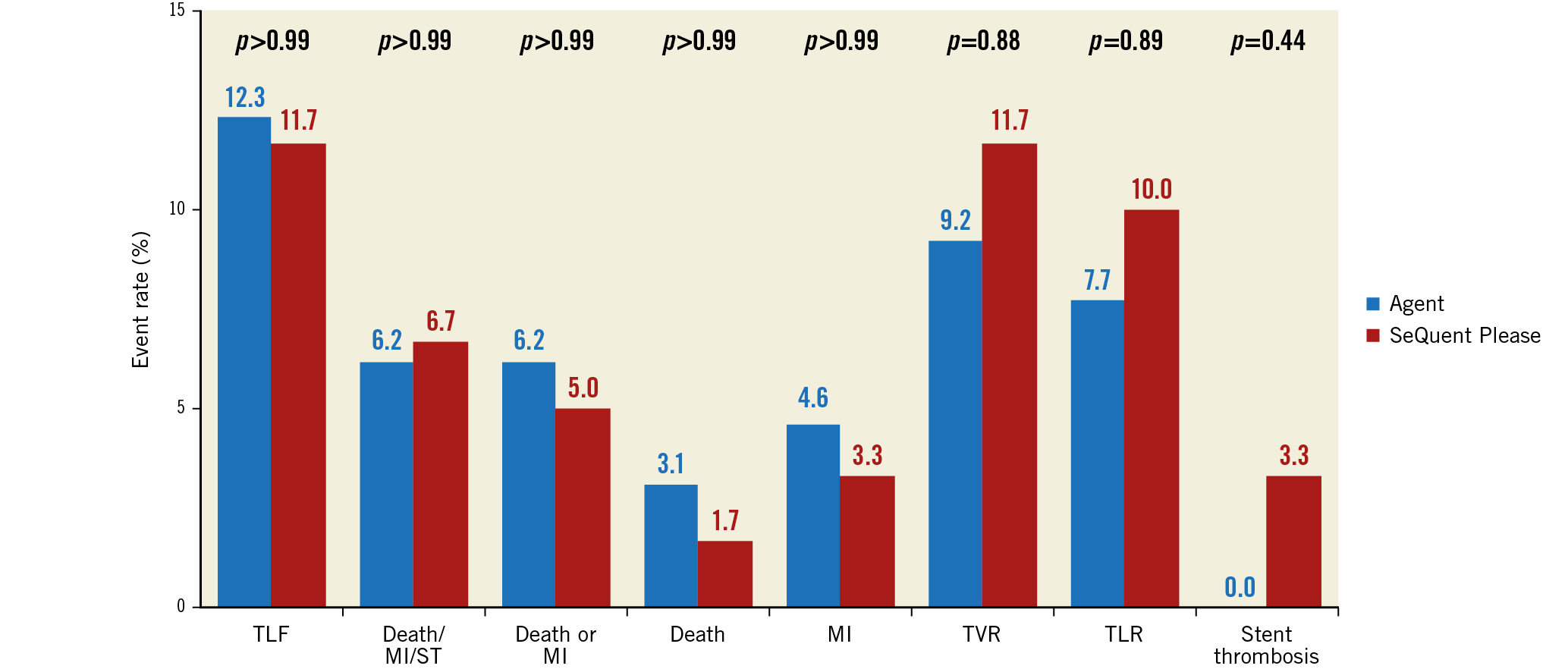
Figure 3. 12-month clinical outcomes. Clinical outcomes at 12 months in the Agent (blue) and SeQuent Please (red) cohorts. MI: myocardial infarction; TLF: target lesion failure; TLR: target lesion revascularisation; TVR: target vessel revascularisation
Discussion
The AGENT ISR study is one of the first head-to-head comparisons of two DCB. In this randomised study, the Agent DCB was non-inferior to the SeQuent Please DCB with respect to six-month in-stent late lumen loss. There were no statistically significant differences in clinical or quality-of-life outcomes between cohorts. No stent thromboses were reported in the Agent cohort at one year. These data support the safety and efficacy of the Agent DCB for treatment of patients with BMS-ISR or DES-ISR.
DCB employ a non-stent-based mechanism to deliver antiproliferative drugs rapidly (<60 seconds) and directly to the vessel wall. The absence of a residual metallic stent and the lack of permanent polymer may reduce the risk of late thrombosis or side branch occlusion18. DCB use in coronary arteries has shown effectiveness in three main situations – in-stent restenosis, small coronary arteries, and bifurcation lesions19. Two network meta-analyses concluded that newer-generation everolimus-eluting stents and DCB were the most effective treatment options for ISR1,20. Although everolimus-eluting stents reduced angiographic and clinical restenosis to a greater extent than DCB, the authors suggested that DCB should be considered for treatment of ISR because the favourable results were obtained without an additional metal layer1.
Baseline characteristics between cohorts were well balanced. There was a numerical increase in preprocedural binary restenosis and lesion calcification in the SeQuent Please group. Calcification may lead to suboptimal dilatation or act as a barrier for drug delivery; however, even though there was a slightly higher post-procedural percent diameter stenosis observed in patients treated with the SeQuent Please DCB, there were no differences between the groups at six-month angiographic follow-up and 12-month clinical follow-up. Drug-coated balloons utilise different solvents and excipients to deliver drug to the vessel wall. Preclinical testing in a porcine model suggested that n-butyryl-tri-n-hexylcitrate (BTHC) and iopromide are the most efficacious excipients3. The Agent DCB utilises a newer technology with an acetyl tri-butyl citrate excipient that allows a reduced drug dose density of 2 μg/mm2 with the aim of reducing vascular toxicity21. Excipient selection and paclitaxel dose may lead to differences in clinical event rates.
A number of other studies have compared different DCB. The RESTORE ISR China randomised controlled trial compared the RESTORE DEB® (3 mg paclitaxel/mm2; shellac-ammonium salt excipient) (Cardionovum GmbH, Bonn, Germany) and the SeQuent Please DCB with DES-ISR in 240 patients regarding the outcome at the one-year follow-up22. In this study, the RESTORE DEB was non-inferior when compared with the SeQuent Please DCB regarding late lumen loss as the primary endpoint. An analysis of 1,129 patients treated with DCB from the Swedish Coronary Angiography and Angioplasty Registry (SCAAR/SWEDEHEART) found that the SeQuent Please DCB had lower restenosis rates than the Elutax (Aachen Resonance, Aachen, Germany) DCB (3.4% vs 12.5%)12. In a retrospective analysis of Pantera Lux- and SeQuent Please-treated patients, Pantera Lux (Biotronik, Bülach, Switzerland) had lower rates of adverse events at three years than SeQuent Please (mainly driven by TLR)23. The combined ISAR-DESIRE 3 and 4 registries did not reveal any distinctions between DCB; diameter stenosis at six to eight months and 12-month clinical outcomes were similar between SeQuent Please and Pantera Lux24. The results of AGENT ISR demonstrate that the Agent DCB performs as effectively as the SeQuent Please DCB and suggest that the Agent DCB may be a useful addition for the treatment of patients with ISR.
Study limitations
Although this study provides important information, there are limitations that have to be considered. Only patients with a single ISR lesion were included, and the study was not powered to detect differences in more clinical endpoints. The results are comparable to those of recent randomised controlled trials; however, the large non-inferiority margin and observed standard deviations as well as possible borderline significance must be taken into account when assessing the present results. Only one-year results are available, which may not reflect differences in long-term clinical outcomes between DCB. Also, this study compared two DCB and did not evaluate other ISR treatments (such as the use of DES).
Conclusions
In this head-to-head comparison of two DCB with different drug formulations, Agent proved to be non-inferior to SeQuent Please for in-stent late lumen loss at six months. Mortality and MI were similar between the groups at 12 months as were quality-of-life outcomes.
|
Impact on daily practice The ideal DCB for treating in-stent restenosis of a previously treated coronary lesion is not yet known. The aim of the randomised, multicentre AGENT ISR study was to evaluate the safety and effectiveness of treating in-stent restenosis with the Agent DCB, which uses a novel excipient and lower antiproliferative drug dose. Outcomes up to one year were compared with those of a standard DCB (SeQuent Please). Agent was non-inferior to SeQuent Please for in-stent late lumen loss at six months. Other angiographic, clinical, and quality-of-life outcomes were similar between the DCB. |
Acknowledgements
The authors thank Kristine Roy, PhD (Boston Scientific Corporation), for manuscript preparation assistance, and Elizabeth Martinson, PhD (KHFI Editorial Office), for additional language editing.
Funding
The trial was supported by Hemoteq AG and Boston Scientific.
Conflict of interest statement
F. Roubille reports other from Abbott, Servier, and Medtronic, grants and other from AstraZeneca, other from Novartis, MSD, Actelion, Thoratec, Sanofi, and Amgen, outside the submitted work. I. Schult is a full-time employee of Hemoteq AG. J. Allocco is a full-time employee and stockholder of Boston Scientific. J. Berland reports grants from Hemoteq. C.W. Hamm reports personal fees from Boston Scientific and Hemoteq during the conduct of the study, and personal fees from Medtronic and Abbott outside the submitted work. The other authors have no conflicts of interest to declare.
Supplementary data
To read the full content of this article, please download the PDF.
